Evaluation of Hypotonia
DEFINITIONS
Muscle tone is the resistance of muscle to passive stretch. Hypotonia is a decreased tone in the limbs, trunk, or other skeletal muscles. With hypotonia, there is decreased resistance to passive movement across a joint.
INTRODUCTION
Hypotonia in a newborn may be caused by a wide variety of conditions. It can occur if there are abnormalities in the peripheral neuromuscular system, the central nervous system (CNS), or both. The severity may vary widely depending on the underlying cause.
HISTORY
A careful history of the pregnancy, labor, and delivery is important in determining possible congenital conditions or antenatal injury. The onset and the quality of fetal movements should be noted. Lack of fetal movements may suggest a myopathy. A history of unusual hyperextension of the neck in utero or breach delivery may suggest a spinal cord injury. Polyhydramnios suggests a fetal swallowing dysfunction, which may be an indicator of brainstem dysgenesis.
A family history of genetic or neuromuscular disorders and mental retardation is critical. A family history of disorders such as myasthenia gravis, muscular dystrophies, or myotonic dystrophies can be useful in determining the etiology of hypotonia in the newborn infant.
The delivery method and course are also important to explore for possible injury to the brain and spinal cord or for hypoxic-ischemic brain injury. Hypoxic-ischemic encephalopathy is 1 of the most common causes of hypotonia in the neonatal intensive care unit intensive care unit.
The gestational age of the infant at delivery is important because most premature infants will display hypotonia. Postterm infants may also have various reasons for being hypotonic.
The neonatal course is also important for determining the temporal nature of the disorder and determining if there are any acquired conditions. Transient hypotonia is common in disorders such as hypermagnesemia (caused by maternal administration of intravenous magnesium) and transient myasthenia of newborns. Neuromuscular disorders and genetic syndromes will have effects on tone that are more lasting.
PHYSICAL EXAMINATION
A careful general and neurologic examination of the newborn may provide clues to the etiology. The general examination should focus on dysmorphic features that could be associated with genetic syndromes (eg, Down syndrome, Prader-Willi syndrome); size and shape of the head; neck and back examination (for any evidence of spinal dysraphisms); extremities (for limb dysgenesis and joint contractures); and organomegaly for storage diseases (eg, Zellweger syndrome).
Hypotonia is assessed by passive range of motion of muscles across joints and functional tests such as the “traction response” test. The infant is pulled by arms from a supine position to a “sitting” position while assessing tone in the shoulder muscle and neck. If the infant has significant hypotonia, there will be a head lag (extension) and poor resistance in the upper limbs (Figure 118-1). The ventral suspension maneuver is also useful to determine the tone in the trunk (Figure 118-1).
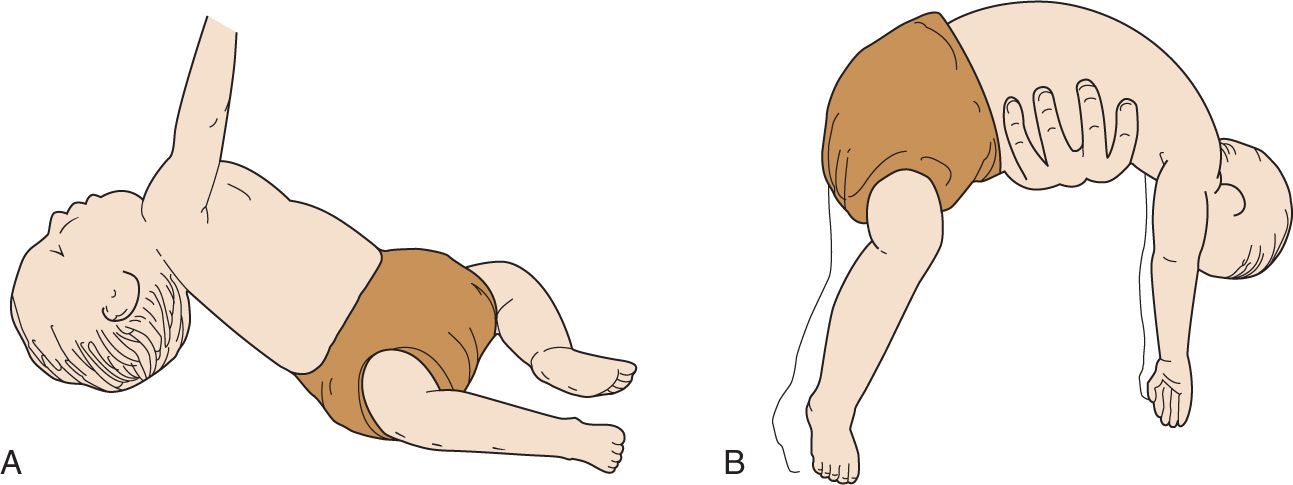
FIGURE 118-1 A, Hypotonic infant pulled into sitting position demonstrates poor head control with lag and lack of flexion of the arm. B, Hypotonic infant supported ventrally in horizontal position demonstrates limbs and head that hang limply. (Reproduced with permission from Dunn and Epstein.1)
The neurologic examination should be focused on eliciting clinical findings of an upper motor neuron vs a peripheral dysfunction. Table 118-1 provides some of the key neurologic examination findings depending on the localization of the dysfunction.
Table 118-1 Physical Examination Findings and Localization
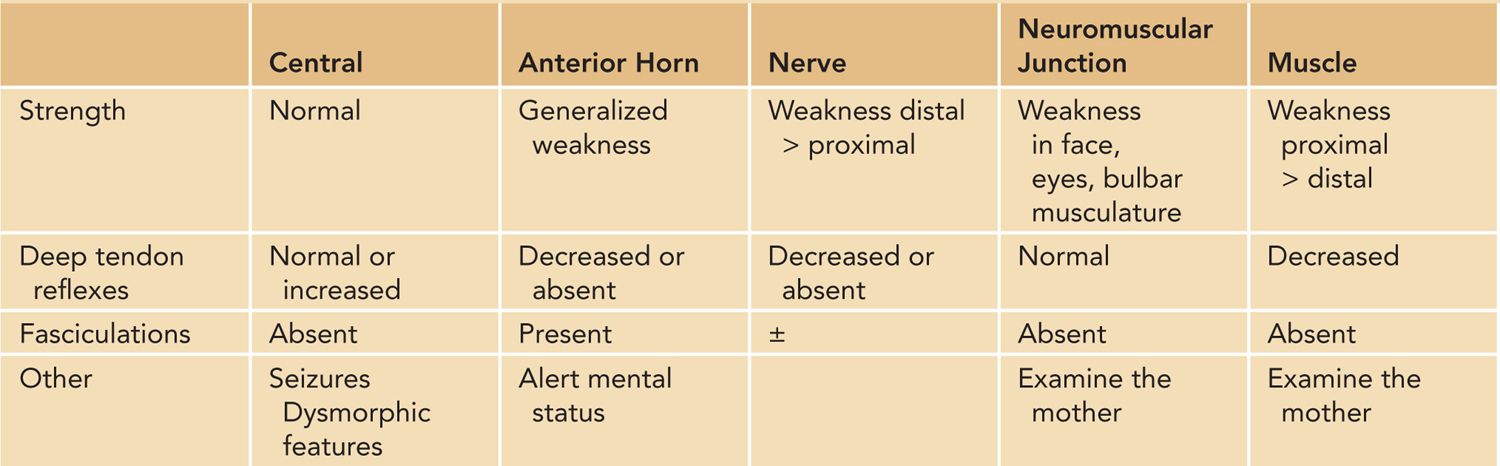
The presence of fasciculations, often most easily detectable on the tongue, would suggest a peripheral nerve or motor neuron disorder. The motor neurons may be located in the anterior horn of the spinal cord or in the brainstem nuclei that control bulbar musculature. The absence of deep tendon reflexes would suggest a peripheral neuropathy or anterior horn cell (AHC) disorder. Preservation of cognitive function in the setting of profound weakness suggests a peripheral process.
Initially, the newborns with upper motor dysfunction have normal deep tendon reflexes and hypotonia. Hyperreflexia and spasticity, hallmarks of upper motor neuron disorders, emerge after the newborn period.
ETIOLOGY
Localization and Differential Diagnosis
Hypotonia can occur if there are abnormalities in the muscle, neuromuscular junction (NMJ), axon/myelin sheath of peripheral nerve, AHCs, long tracts of the spinal cord, and the CNS (Figure 118-2). Table 118-2 provides various etiologies categorized by neurological localization. Hypotonia may also occur outside the nervous system and the motor unit. Table 118-3 provides various nonneurological etiologies.
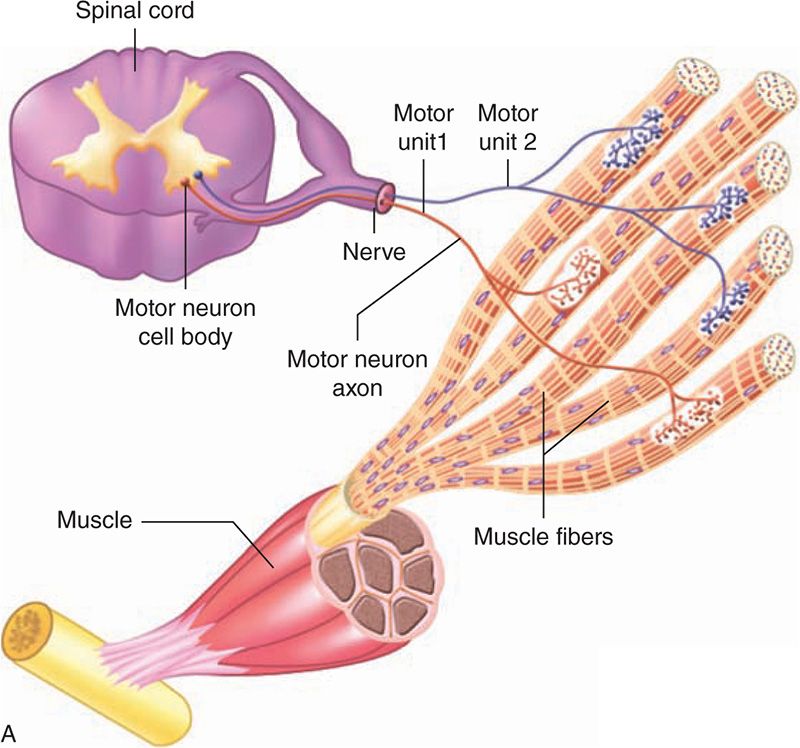
FIGURE 118-2 The motor unit is composed of the anterior horn cell, peripheral nerve, neuromuscular junction, and muscle.
Table 118-2 Differential Diagnosis of Hypotonia (Neurological Cause)
• Central
– Central nervous system
» Hypoxic-ischemic encephalopathy
» Intracranial hemorrhage
» Chromosomal or gene abnormalities (Down syndrome, Prader-Willi syndrome, Angelman syndrome, William syndrome)
» Cerebral malformations
» Infections (sepsis, meningoencephalitis, TORCH, HIV)
» Peroxisomal disorders (Zellweger, neonatal adrenoleukodystrophy)
» Toxic/metabolic encephalopathy (benzodiazepine, hypothyroidism, kernicterus)
– Spinal cord
» Birth trauma
» Spinal cord dysraphisms (spina bifida, myelomeningocele, syringomyelia)
– Anterior horn cell
» Spinal muscular atrophy (Werdnig-Hoffman disease)
– Benign familial hypotonia
• Peripheral
– Peripheral nerve
» Hereditary motor and sensory neuropathy
» Congenital hypomyelination neuropathy
» Infantile neuroaxonal dystrophy
» Familial dysautonomia
» Guillain-Barré syndrome
– Neuromuscular junction
» Myasthenic syndromes
• Transient neonatal myasthenia
• Congenital myasthenia syndromes (channelopathies)
• Familial infantile myasthenia
» Infantile botulism
» Magnesium toxicity
– Muscle
» Structurally specific congenital myopathies
• Nemaline (rod) myopathy
• Central core myopathy
• Myotubular myopathy
• Congenital fiber-type disproportion
» Muscular dystrophies
• Myotonic dystrophy
• Infantile fascioscapulohumeral dystrophy
• Congenital muscular dystrophies (including merosin deficiency)
» Myopathies with biochemical cause
• Glycogen storage diseases (Pompe disease)
• Lipid myopathies
• Mitochondrial myopathies
• Carnitine palmitoyltransferase deficiency type 2 (neonatal lethal form)
• Combined central and peripheral
– Dystroglycanopathies
» Walker-Warburg syndrome
» Fukuyama type congenital muscular dystrophy
» Muscle-eye-brain disease
– Congenital disorders of glycosylation
– Canavan disease
– Mitochondrial encephalomyopathies
– Congenital Pelizaeus-Merzbacher disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


