Creatinine is a reflection of body muscle mass. Because accepted ranges of normal Ccr are based on adult parameters, correction for size is needed to determine normal ranges in children. Clearance is corrected to a standard body surface area of 1.73 m2 in the formula:
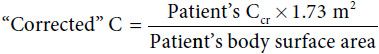
Although 80–125 mL/min/1.73 m2 is the normal range for Ccr, estimates at the lower end of this range may indicate problems.
The Schwartz formula is a reliable formula for quick approximation of Ccr based on plasma creatinine level and length in centimeters:
Ccr (mL/min/1.73 m2) = k × height (cm)/Pcr (mg/dL)
where k is a constant: 0.45 for infants 1–52 weeks old, 0.55 for children 1–13 years old, 0.55 for females 1–18 years old, and 0.7 for males 13–18 years old.
Urine Concentrating Ability
Inability to concentrate urine causes polyuria, polydipsia, or enuresis and is often the first sign of chronic renal failure, and, in some cases, raises the possibility of diabetes insipidus. The first morning void should be concentrated (specific gravity 1.020 or higher), presuming cessation of drinking anything through the night. Thus, determination of the specific gravity of a first morning void is an easy and helpful test of the kidney’s concentrating ability.
Urinalysis
Commercially available dipsticks can be used to screen the urine for blood leukocytes, nitrites, protein, and specific gravity and to approximate the urine pH. Positive results for blood should always be confirmed by microscopy, which is also the only way to determine if there is significant crystalluria. Significant proteinuria (> 150 mg/dL) detected by dipstick should be confirmed by quantitation, either with a 24-hour urine collection or by the protein/creatinine ratio of a random specimen.
In children with asymptomatic hematuria or proteinuria, the search for renal origins will yield the most results. Isolated proteinuria may also reflect urologic abnormalities, benign excretion, or glomerular alterations. RBC casts suggest glomerulonephritis (GN), but the absence of casts does not rule out this disease. Anatomic abnormalities such as cystic disease may also cause hematuria.
Benign hematuria, including benign familial hematuria, is diagnosed by exclusion. In this group are children whose hematuria is caused by asymptomatic hypercalciuria. Figure 24–1 suggests an approach to the renal workup of hematuria. GN is discussed in more detail later in this chapter.
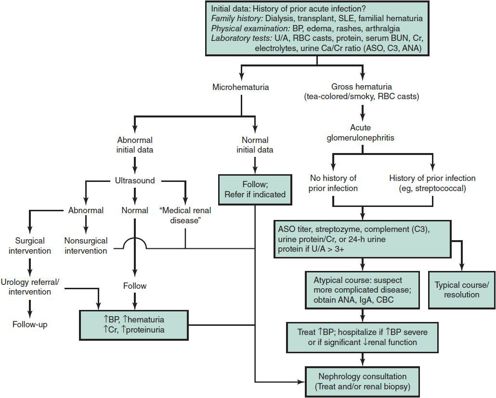
 Figure 24–1. Approach to the renal workup of hematuria. (Exclude UTI, lithiasis, trauma, bleeding disorders, sickle cell disease.) Complement is depressed in acute poststreptococcal type of glomerulonephritis (about 30 days), chronic glomerulonephritis (persistent), and lupus. ANA, antinuclear antibody; ASO, antistreptolysin antibody; BP, blood pressure; BUN, blood urea nitrogen; C3, complement; Ca, calcium; CBC, complete blood count; Cr, creatinine; IgA, immunoglobulin A; RBC, red blood cell; SLE, systemic lupus erythematosus; U/A, urinalysis.
Figure 24–1. Approach to the renal workup of hematuria. (Exclude UTI, lithiasis, trauma, bleeding disorders, sickle cell disease.) Complement is depressed in acute poststreptococcal type of glomerulonephritis (about 30 days), chronic glomerulonephritis (persistent), and lupus. ANA, antinuclear antibody; ASO, antistreptolysin antibody; BP, blood pressure; BUN, blood urea nitrogen; C3, complement; Ca, calcium; CBC, complete blood count; Cr, creatinine; IgA, immunoglobulin A; RBC, red blood cell; SLE, systemic lupus erythematosus; U/A, urinalysis.
Combined proteinuria and hematuria is characteristic of more significant glomerular disease. Quantitation of proteinuria is customarily accomplished by a timed collection (eg, over a 24-hour period). However, given the frequency of errors in collection in the pediatric population, the degree of proteinuria may be estimated by the ratio of protein mg/dL/creatinine mg/dL in a random urine sample. A protein/creatinine ratio above 0.2 is abnormal. If the laboratory reports this as mg protein/gram of creatinine, normal is 200 or less.
In the evaluation of asymptomatic proteinuria, orthostatic or postural proteinuria should be ruled out. This can be accomplished simply by comparing the protein/creatinine ratio of urine formed in the supine position (the first morning void accumulated in the bladder while sleeping) to a sample obtained during daily ambulation. If the “supine” sample is normal and proteinuria is occurring only during upright posture, this demonstrates postural (benign) proteinuria. If both samples are abnormal, proteinuria would be considered “persistent.”
An approach to the workup of isolated proteinuria, including nephrotic syndrome, is shown in Figure 24–2. Note that corticosteroid therapy is included in the algorithm because this may be initiated for nephrotic syndrome prior to referral. Other renal lesions with proteinuric manifestations are discussed later in this chapter.
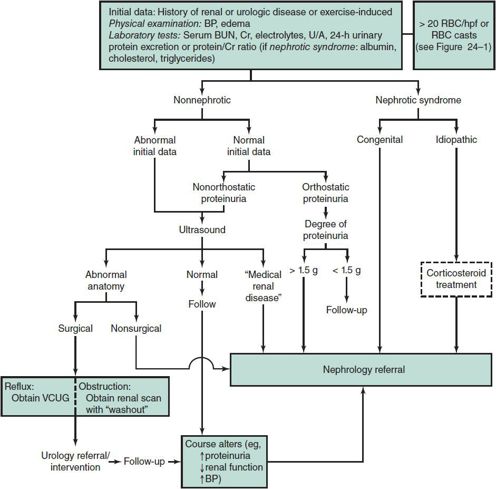
 Figure 24–2. Approach to the workup of isolated proteinuria. BP, blood pressure; Cr, creatinine; hpf, high-power field; RBC, red blood cell; U/A, urinalysis; VCUG, voiding cystourethrogram. Rules out benign postural proteinuria with urine protein/creatine ratio of first morning void (recumbent urine) versus day void (upright). Will normalize within a month in poststreptococcal glomerulonephritis.
Figure 24–2. Approach to the workup of isolated proteinuria. BP, blood pressure; Cr, creatinine; hpf, high-power field; RBC, red blood cell; U/A, urinalysis; VCUG, voiding cystourethrogram. Rules out benign postural proteinuria with urine protein/creatine ratio of first morning void (recumbent urine) versus day void (upright). Will normalize within a month in poststreptococcal glomerulonephritis.
Special Tests of Renal Function
Measurements of urinary sodium, creatinine, and osmolality are useful in differentiating prerenal from renal causes of renal insufficiency, such as acute tubular necrosis. Prolonged underperfusion causes varying increases in serum creatinine and blood urea nitrogen (BUN) concentrations, prompting the need to differentiate between this state and acute tubular necrosis (see section Acute Renal Failure). The physiologic response to decreased renal perfusion is decreased urinary output, increased urine osmolality, increased urinary solutes (eg, creatinine), and decreased urinary sodium (usually < 20 mEq/L).
The presence of certain substances in urine may suggest tubular dysfunction. For example, urine glucose should be less than 5 mg/dL. Hyperphosphaturia occurs with significant tubular abnormalities (eg, Fanconi syndrome). Measurement of the phosphate concentration of a 24-hour urine specimen and evaluation of tubular reabsorption of phosphorus (TRP) will help document renal tubular diseases as well as hyperparathyroid states. TRP (expressed as percentage of reabsorption) is calculated as follows:
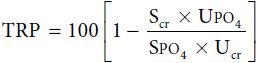
where Scr = serum creatinine; Ucr = urine creatinine; SPO4 = serum phosphate; and UPO4 = urine phosphate. All values for creatinine and phosphate are expressed in milligrams per deciliter for purposes of calculation. A TRP value of 80% or more is considered normal, although it depends somewhat on the value of SPO4.
The urinary excretion of amino acids in generalized tubular disease reflects a quantitative increase rather than a qualitative change. Diseases affecting proximal tubular reabsorption of bicarbonate—including isolated renal tubular acidosis (RTA), Fanconi syndrome (which occurs in diseases such as cystinosis), and chronic renal failure—are discussed later in the chapter.
LABORATORY EVALUATION OF IMMUNOLOGIC FUNCTION
Many parenchymal renal diseases are thought to have immune causation, although the mechanisms are largely unknown. Examples include (1) deposition of circulating antigen-antibody complexes that are directly injurious or incite injurious responses and (2) formation of antibody directed against the glomerular basement membrane (rare in children).
C3 and C4 complement components should be measured when immune-mediated renal injury or chronic GN is suspected. Where clinically indicated, antinuclear antibodies, hepatitis B surface antigen, and rheumatoid factor should be obtained. In rare cases, cold-precipitable proteins (cryoglobulins), C3 “nephritic” factor, or antiglomerular basement membrane (anti-GBM) antibody measurements may help confirm a specific diagnosis. At some point in the workup, the diagnosis may be supported or confirmed by histologic examination of renal tissue.
RADIOGRAPHIC EVALUATION
Renal ultrasonography is a useful noninvasive tool for evaluating renal parenchymal disease, urinary tract abnormalities, or renal blood flow. Excretory urography is used to assess the anatomy and function of the kidneys, collecting system, and bladder. Radioisotope studies provide information about renal anatomy, blood flow, and integrity and function of the glomerular, tubular, and collecting systems. Renal stones are best seen by computed tomography.
Voiding cystourethrography or cystoscopy is indicated when vesicoureteral reflux (VUR) or bladder outlet obstruction is suspected. Cystoscopy is rarely useful in the evaluation of asymptomatic hematuria or proteinuria in children.
Renal arteriography or venography is indicated to define vascular abnormalities (eg, renal artery stenosis) prior to surgical intervention or transluminal angiography. Less invasive measures such as ultrasonography and Doppler studies can demonstrate renal blood flow or thromboses. More specific identification of stenoses of the renal artery is accomplished by magnetic resonance arteriography.
RENAL BIOPSY
Histologic information is valuable for diagnosis, to guide treatment, and to inform prognosis. Satisfactory evaluation of renal tissue requires examination by light, immunofluorescence, and electron microscopy. The need for a renal biopsy should be determined by a pediatric nephrologist.
CONGENITAL ANOMALIES OF THE URINARY TRACT
RENAL PARENCHYMAL ANOMALIES
About 10% of children have congenital anomalies of the genitourinary tract, which range in severity from asymptomatic to lethal. Some asymptomatic abnormalities may have significant complications. For example, patients with “horseshoe” kidney (kidneys fused in their lower poles), although not representing renal parenchymal disease or reduction in kidney function, have a higher incidence of renal calculi. Unilateral agenesis or multicystic dysplasia is usually accompanied by compensatory hypertrophy of the contralateral kidney and thus should be compatible with normal renal function, requiring no specific nephrology referral or follow-up. Supernumerary and ectopic kidneys are usually of no significance. Abnormal genitourinary tract development is associated with varying degrees of renal dysgenesis and dysfunction ranging from mild to severe; an example of the latter is in utero bilateral renal agenesis, which is associated with severe oligohydramnios, pulmonary hypoplasia, abnormal (Potter) facies, and perinatal death.
1. Renal Dysgenesis
Renal dysgenesis is a spectrum of anomalies. In simple hypoplasia, which may be unilateral or bilateral, the affected kidneys are smaller than normal. In some forms of dysgenesis, immature, undifferentiated renal tissue persists. In some situations, the number of normal nephrons is insufficient to sustain life once the child reaches a critical body size. The lack of adequate renal tissue may not be readily discernible in the newborn period in the presence of normal urine production. Often, discovery of renal insufficiency in an infant is coincident with blood work drawn for other purposes showing an elevated serum creatinine.
Other forms of renal dysgenesis include oligomeganephronia (characterized by the presence of only a few large glomeruli) and the cystic dysplasias (characterized by the presence of renal cysts). This group includes microcystic disease (congenital nephrosis). A simple cyst within a kidney, different from either autosomal recessive or dominant polycystic kidney disease, is clinically unimportant. An entire kidney lost to multicystic development with concomitant hypertrophy and normal function of the contralateral side should also be of no clinical consequence.
2. Polycystic Kidney Disease
Both forms of polycystic kidney disease (autosomal dominant [ADPKD] or recessive [ARPKD]) are increasingly diagnosed by prenatal ultrasound. In its most severe form (ARPKD), the cystic kidneys are nonfunctional in utero, and, therefore, newborns can have Potter facies and other complications of oligohydramnios. In infancy and childhood, kidney enlargement by cysts may initially be recognized by abdominal palpation of renal masses. Hypertension is an early problem in ARPKD. The rate of the progression of renal insufficiency varies, as does growth failure, other complications of chronic renal failure, and early development of end-stage renal disease. In ADPKD, two genes, ADPKD1 and ADPKD2, account for 80% and 10% of cases, respectively. Susceptibility of family members is detected by gene linkage studies. Renal ultrasound identifies cysts in about 80% of affected children by age 5 years. Children with this diagnosis need monitoring for the development and treatment of hypertension, which usually develops in the teenage years. These patients would not be expected to develop renal insufficiency, if any, until later in adult years.
3. Medullary Cystic Disease (Juvenile Nephronophthisis)
Medullary cystic disease is characterized by cysts of varying sizes in the renal medulla with tubular and interstitial nephritis. Children present with renal failure and signs of tubular dysfunction (decreased concentrating ability, Fanconi syndrome). This lesion should not be confused with medullary sponge kidney (renal tubular ectasia), a frequently asymptomatic disease occurring in adults.
DISTAL URINARY TRACT ANOMALIES
1. Obstructive Uropathy
Obstruction at the ureteropelvic junction may be the result of intrinsic muscle abnormalities, aberrant vessels, or fibrous bands. The lesion can cause hydronephrosis and usually presents as an abdominal mass in the newborn. Obstruction can occur in other parts of the ureter, especially at its entrance into the bladder, causing proximal hydroureter and hydronephrosis. Renal radionuclide scan with furosemide “wash-out” will reveal or rule out obstruction as the cause of the hydronephrosis. Whether intrinsic or extrinsic, urinary tract obstruction should be relieved surgically as soon as possible to minimize damage to the kidneys.
Severe bladder malformations such as exstrophy are clinically obvious and a surgical challenge. More subtle—but urgent in terms of diagnosis—is obstruction of urine flow from vestigial posterior urethral valves. This anomaly, which occurs almost exclusively in males, usually presents in newborns with anuria or a poor voiding stream secondary to severe obstruction of urine flow. The kidneys and bladder may be easily palpable. Leakage (ureteric perforation, although rare) proximal to the obstruction may produce urinary ascites. Surgical drainage of urine is urgently required to prevent irreversible damage.
Prune belly syndrome is an association of urinary tract anomalies with cryptorchidism and absent abdominal musculature. Although complex anomalies, especially renal dysplasia, usually cause early death or the need for dialysis or transplantation, some patients have lived into the third decade with varying degrees of renal insufficiency. Timely urinary diversion is essential to sustain renal function.
Other complex malformations and external genital anomalies such as hypospadias are beyond the scope of this text. The challenge presented by urologic abnormalities resulting in severe compromise and destruction of renal tissue is to preserve all remaining renal function and treat the complications of progressive chronic renal failure. Involvement of a specialist in pediatric urology in early management is essential.
2. Reflux Nephropathy
The retrograde flow of urine from the bladder into the ureter (vesicoureteral reflux) may cause renal scarring and subsequent renal insufficiency or hypertension, or both, especially in the presence of UTI. A finding of hydronephrosis on renal ultrasound is suggestive of vesicoureteral reflux. Its presence can be confirmed or eliminated by a voiding cystourethrogram, which would also be obtained to rule out reflux in the evaluation of UTI. Low-grade reflux may resolve in the absence of infection, in which case antibiotic prophylaxis (advised with any degree of reflux) is undertaken while awaiting signs of spontaneous resolution. Surgery may be required for chronic severe reflux.
HEMATURIA & GLOMERULAR DISEASE
MICROHEMATURIA
Children with painful hematuria should be investigated for UTI or direct injury to the urinary tract. Dysuria is common in cystitis or urethritis; associated back pain suggests the possibility of pyelonephritis; colicky flank pain may indicate the passage of a stone. Bright red blood or clots in the urine are associated with bleeding disorders, trauma, and arteriovenous malformations. Abdominal masses suggest the presence of urinary tract obstruction, cystic disease, or tumors of the renal or perirenal structures.
Asymptomatic hematuria is a challenge because clinical and diagnostic data are required to decide whether to refer the child to a nephrologist. The diagnosis of hematuria should not rely solely on a urine “dipstick” evaluation, but should be verified by a microscopic RBC count. Ruling out hypercalciuria as a cause of hematuria by a random urine calcium/creatinine ratio is one of the initial steps in the evaluation of hematuria. A value above 0.2 requires verification with a 24-hour collection. Hypercalciuria is excretion of calcium in excess of 4 mg/kg/d. Figure 24–1 delineates the outpatient approach to renal hematuria. The concern regarding the differential diagnosis is the possible presence of glomerular disease.
GLOMERULONEPHRITIS
The various types of glomerulonephritis (GN) have similar manifestations. Table 24–1 lists the most commonly encountered disorders in the differential diagnosis of childhood GN, including their clinical and histopathologic abnormalities. Severe glomerular histopathologic and clinical entities, such as anti-GBM antibody disease (Goodpasture syndrome), Wegener granulomatosis, and idiopathic, rapidly progressive GN, may be considered in the differential diagnosis of acute GN, but these disorders are exceedingly rare in children.
Table 24–1. Glomerular diseases encountered in childhood.
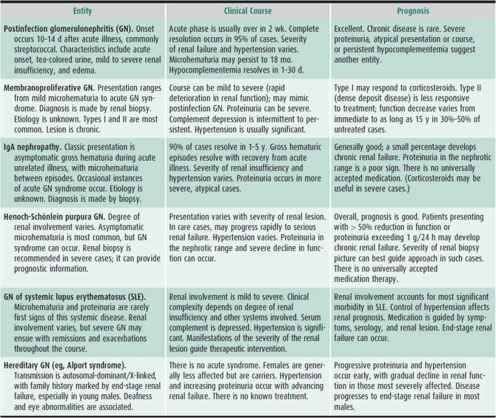
1. Acute Poststreptococcal Glomerulonephritis
The diagnosis of acute poststreptoccocal glomerulonephritis is supported by a recent history (7–14 days previously) of group A β-hemolytic streptococcal infection, typically involving the pharynx or skin. If a positive culture is not available, recent infection may be supported by an elevated antistreptolysin O titer or by high titers of other antistreptococcal antibodies. Other infections can cause similar glomerular injury; thus, “postinfection” glomerulonephritis (GN) may be a better term for this type of acute glomerulonephritis (AGN). In most cases, recovery is expected and usually complete within weeks. If the diagnosis is in question, or if the renal function of a patient with postinfection GN progressively deteriorates, a renal biopsy should be performed and treatment with corticosteroids initiated.
The clinical presentation of GN is gross hematuria generally accompanied by varying degrees of increase in serum creatinine, edema, and hypertension. Urine may be coffee-colored or tea-colored. Microscopic examination of urine reveals RBCs too numerous to count. Microscopy may reveal RBC casts. If present, these are diagnostic of GN, but their absence does not exclude the diagnosis. Edema is often seen (periorbital, facial, extremities), caused by sodium and water retention resulting from alteration in glomerular function. Symptoms are usually nonspecific. In cases accompanied by hypertension (a common finding), headache may be present. Fever is uncommon. Severe glomerular injury (which usually occurs in severe, acute presentations of the more chronic or destructive forms of GN) may be accompanied by massive proteinuria (nephrotic syndrome), anasarca, ascites, and severe compromise of renal function.
Typical poststreptococcal GN has no specific treatment. Antibiotic therapy is indicated if an infection is still present. Disturbances in renal function and resulting hypertension may require close monitoring, reduction in salt intake, diuretics, or other antihypertensive drugs. In severe cases of renal failure, hemodialysis or peritoneal dialysis may be necessary. Corticosteroids may also be administered in an attempt to influence the course of the GN.
The acute abnormalities generally resolve in 2–3 weeks. Low levels of serum complement (C3) may normalize as early as 24 hours or as late as 30 days after onset. Other complement-consuming glomerulonephritides include membranoproliferative GN (chronic GN with persistent complement depression) and lupus GN. In poststreptococcal GN, although microscopic hematuria may persist for as long as a year, 85% of children recover completely. Persistent deterioration in renal function, urinary abnormalities beyond 18 months, persistent hypocomplementemia, and nephrotic syndrome are ominous signs. If any of these is present, a renal biopsy is indicated.
2. IgA Nephropathy
When asymptomatic gross hematuria appears to accompany a minor acute febrile illness or other stressful occurrence, the diagnosis of IgA nephropathy may be entertained. In contrast to postinfection GN, IgA nephropathy is not associated with prior streptococcal infection, complement is not depressed, and in 50% of cases, serum immunoglobin A is elevated. Often there are no associated symptoms or signs. Gross hematuria resolves within days, and there are no serious sequelae in 85% of cases. Treatment is not indicated, and the prognosis is good in most cases. Prognosis is guarded, however, if severe proteinuria, hypertension, or renal insufficiency is present or develops. In such instances, although no treatment is universally accepted, corticosteroids and other immunosuppressive drugs are used. Omega-3 fatty acids from fish oils are thought to be helpful.
3. Henoch-Schönlein Purpura
The diagnosis of Henoch-Schönlein purpura rests on the presence of a typical maculopapular and purpuric rash found primarily, but not exclusively, on the dorsal surfaces of the lower extremities and buttocks. Most children have abdominal pain, and bloody diarrhea may be present. Joint pain is common, and, depending on the extent of renal involvement, hypertension may be present. Joint and abdominal pain responds to treatment with corticosteroids. Renal involvement ranges from mild GN with microhematuria to severe GN and varying degrees of renal insufficiency. GN with massive proteinuria and renal insufficiency carries a poor prognosis. Twenty percent of such cases result in end-stage renal failure. There is no universally accepted treatment, but corticosteroids are often administered (see Chapter 30).
4. Membranoproliferative Glomerulonephritis
The most common “chronic” form of GN in childhood is membranoproliferative GN. The diagnosis is established from the histologic appearance of the glomeruli on biopsy tissue. There are two major histologic types of membranoproliferative GN. Clinically, type II carries the worse prognosis, as end-stage renal failure develops in most cases. Type I more often responds to treatment with corticosteroids. C3 is depressed (in both types) and may be useful as a marker of response to treatment.
5. Lupus Glomerulonephritis
The diagnosis of systemic lupus erythematosus (SLE) is based on its numerous clinical features and abnormal laboratory findings that include a positive antinuclear antibody test, depressed serum complement, and increased serum double-stranded DNA. Renal involvement is indicated by varying degrees of hematuria and proteinuria. More severe cases are accompanied by renal insufficiency and hypertension. Significant renal involvement requires treatment with various combinations of immunosuppressive drugs including prednisone (as a primary drug), azathioprine, cyclophosphamide, mycophenalate, tacrolimus, and rituximab, a monoclonal antibody against the B-cell surface antigen CD20. End-stage renal failure develops in 10%–15% of patients with childhood SLE.
6. Hereditary Glomerulonephritis
The most commonly encountered hereditary GN is Alport syndrome, characterized by hearing loss and GN, occurring predominantly in males. It is a chronic form of GN and thus does not present with the clinical features typically seen in patients with acute processes. A family history is generally present, but there is a spontaneous mutation rate of about 18%. In individuals with the progressive form of GN, end-stage renal failure occurs, usually in the second to third decade of life. Although currently there is no treatment for this disorder, careful management of associated hypertension may slow the process.
Ahn SY, Ingulli E: Acute poststreptococcal glomerulonephritis: An update. Curr Opin Pediatr 2008;20:157–162 [PMID: 18332711].
Sanders JT, Wyatt RJ: IgA nephropathy and Henoch-Schönlein purpura nephritis. Curr Opin Pediatr 2008;20:163–170 [PMID: 18332712].
ACUTE INTERSTITIAL NEPHRITIS
Acute interstitial nephritis is characterized by diffuse or focal inflammation and edema of the renal interstitium and secondary involvement of the tubules. The condition is most commonly drug related (eg, β-lactam–containing antibiotics, such as methicillin).
Fever, rigor, abdominal or flank pain, and rashes may occur in drug-associated cases. Urinalysis usually reveals leukocyturia and hematuria. Hansel staining of the urinary sediment often demonstrates eosinophils. The inflammation can cause significant deterioration of renal function. If the diagnosis is unclear because of the absence of a history of drug or toxin exposure or the absence of eosinophils in the urine, a renal biopsy may be performed to demonstrate the characteristic tubular and interstitial inflammation. Immediate identification and removal of the causative agent is imperative and may be all that is necessary. Treatment with corticosteroids is helpful in patients with progressive renal insufficiency or nephrotic syndrome. Severe renal failure requires supportive dialysis.
Gonzalez E et al: Early steroid treatment improves the recovery of renal function in patients with drug-induced acute interstitial nephritis. Kidney Int 2008;73:940–946 [PMID: 18185501].
PROTEINURIA & RENAL DISEASE
Urine is rarely completely protein-free, but the average excretion is well below 150 mg/24 h. Small increases in urinary protein can accompany febrile illnesses or exertion and in some cases occur while in the upright posture.
An algorithm for investigation of isolated proteinuria is presented in Figure 24–2. In idiopathic nephrotic syndrome without associated features of GN, treatment with corticosteroids may be initiated. Nephrologic advice or follow-up should be sought, especially in patients with difficult or frequently relapsing unexplained proteinuria.
CONGENITAL NEPHROSIS
Congenital nephrosis is a rare autosomal recessive disorder. The kidneys are pale and large and may show microcystic dilations (microcystic disease) of the proximal tubules and glomerular abnormalities, including proliferation, crescent formation, and thickening of capillary walls. The pathogenesis is not well understood.
Infants with congenital nephrosis commonly have low birth weight, a large placenta, wide cranial sutures, and delayed ossification. Mild edema may be seen after the first few weeks of life. Anasarca follows, and the abdomen can become greatly distended by ascites. Massive proteinuria associated with typical-appearing nephrotic syndrome and hyperlipidemia is the rule. Hematuria is common. If the patient lives long enough, progressive renal failure occurs. Most affected infants succumb to infections by a few months of life.
Treatment prior to dialysis and transplantation has little to offer other than nutritional support and management of the chronic renal failure.
IDIOPATHIC NEPHROTIC SYNDROME OF CHILDHOOD (MINIMAL CHANGE DISEASE)
Nephrotic syndrome is characterized by proteinuria, hypoproteinemia, edema, and hyperlipidemia. It may occur as a result of any form of glomerular disease and may rarely be associated with a several extrarenal conditions. In young children, the disease usually takes the form of idiopathic nephrotic syndrome of childhood (nil disease, lipoid nephrosis, minimal change disease), which has characteristic clinical and laboratory findings, but no well-understood cause.
 Clinical Findings
Clinical Findings
Affected patients are generally younger than age 6 years at onset. Typically, periorbital swelling and oliguria are noted, often following an influenza-like syndrome. Within a few days, increasing edema—even anasarca—becomes evident. Most children have few complaints other than vague malaise or abdominal pain. With significant “third spacing” of plasma volume, however, some children may present with hypotension. With marked edema, dyspnea due to pleural effusions may also occur.
Despite heavy proteinuria, the urine sediment is usually normal, although microscopic hematuria may be present. Plasma albumin concentration is low, and lipid levels increased. When azotemia occurs, it is usually secondary to intravascular volume depletion.
Glomerular morphology is unremarkable except for fusion of foot processes of the glomerular basement membrane. This nonspecific finding is associated with many proteinuric states.
 Complications
Complications
Infections (eg, peritonitis) sometimes occur, and Streptococcus pneumoniae is frequently the cause. Hypercoagulability may be present, and thromboembolic phenomena are commonly reported. Hypertension can be noted, and renal insufficiency can result from decreased renal perfusion.
 Treatment & Prognosis
Treatment & Prognosis
As soon as the diagnosis of idiopathic nephrotic syndrome is made, corticosteroid treatment should be started. Prednisone, 2 mg/kg/d (maximum, 60 mg/d), is given for 6 weeks as a single daily dose. The same dose is then administered on an alternate-day schedule for 6 weeks; thereafter, the dose is tapered gradually and discontinued over the ensuing 2 months. The goal of this regimen is the disappearance of proteinuria. If remission is not achieved during the initial phase of corticosteroid treatment, additional nephrologic consultation should be obtained. If remission is achieved, only to be followed by relapse, the treatment course may be repeated. A renal biopsy is often considered when there is little or no response to treatment. One should take into account that the histologic findings may not alter the treatment plan, which is designed to eliminate the nephrotic syndrome regardless of underlying renal histology.
Unless the edema causes symptoms such as respiratory compromise due to ascites, diuretics should be used with extreme care. Patients may have decreased circulating volume and are also at risk for venous thrombosis. Careful restoration of compromised circulating volume with intravenous 25% albumin infusion and administration of a diuretic such as furosemide is helpful in mobilizing edema. Infections such as peritonitis should be treated promptly to reduce morbidity. Immunization with pneumococcal conjugate and polysaccharide vaccines is advised.
A favorable response of proteinuria to corticosteroids and subsequent favorable response during relapse suggests a good prognosis. Failure to respond or early relapse usually heralds a prolonged series of relapses. This not only may indicate the presence of more serious nephropathy, but presents a challenge in choosing future therapy for those either severely corticosteroid “dependent” and/or in danger of increasing steroid side effects. Historically, chlorambucil or cyclophosphamide drug therapy added to corticosteroid treatment have been utilized in an attempt to achieve corticosteroid discontinuance while maintaining remission. Such drugs are often used effectively, but only in children who respond well to corticosteroids in the first place. Because of potential significant side effects associated with these drugs, tacrolimus or cyclosporine is now added instead for the treatment of steroid dependent cases. Increasing reports and experience suggest that cases in which nephrotic syndrome is poorly responsive to or “dependent” upon corticosteroids, even with an added agent such as tacrolimus, may respond to rituximab. Patients who do not respond to corticosteroids or who relapse frequently should be referred to a pediatric nephrologist, if such referral was not made earlier in the course.
FOCAL GLOMERULAR SCLEROSIS
Focal glomerular sclerosis is one cause of corticosteroid-resistant or frequent relapsing nephrotic syndrome. The cause is unknown. The diagnosis is made by renal biopsy, which shows normal-appearing glomeruli as well as some partially or completely sclerosed glomeruli. The lesion has serious prognostic implications because as many as 15%–20% of cases can progress to end-stage renal failure. The response to corticosteroid treatment is variable. In difficult cases, especially when prolonged use of steroids is resulting in significant undesirable side effects, other immunosuppressive agents such as cyclosporin A or tacrolimus have been used in addition to corticosteroids to try to achieve longer remission with corticosteroid discontinuance. Recurrence of focal glomerular sclerosis resulting in nephrotic syndrome may occur after renal transplantation. The recurrence is usually treated with plasmapheresis and/or rituximab; the latter agent is also showing encouraging utility in treating the nephrotic syndrome of membranous or mesangial nephropathy as well as refractory nephrotic syndrome associated with other forms of glomerular disease or vasculitis.
MESANGIAL NEPHROPATHY (MESANGIAL GLOMERULONEPHRITIS)
Mesangial nephropathy is another form of corticosteroid-resistant nephrotic syndrome. The renal biopsy shows a distinct increase in the mesangial matrix of the glomeruli. Very often the expanded mesangium contains deposits of IgM demonstrable on immunofluorescent staining. The cause is unknown. Corticosteroid therapy may induce remission, but relapses are common. Choices for treating this type of nephrotic syndrome are the same as noted earlier.
MEMBRANOUS NEPHROPATHY (MEMBRANOUS GLOMERULONEPHRITIS)
Although largely idiopathic in nature, membranous nephropathy can be found in association with hepatitis B antigenemia, SLE, congenital and secondary syphilis, renal vein thrombosis; with immunologic disorders such as autoimmune thyroiditis; and with administration of drugs such as penicillamine. The pathogenesis is unknown, but the glomerular lesion is thought to be the result of prolonged deposition of circulating antigen-antibody complexes.
The onset of membranous nephropathy may be insidious or may resemble that of idiopathic nephrotic syndrome of childhood (see earlier section). It occurs more often in older children and adults. The proteinuria of membranous nephropathy responds poorly to corticosteroid therapy, although low-dose corticosteroid therapy may reduce or delay development of chronic renal insufficiency. The diagnosis is made by renal biopsy.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


