Juvenile Dermatomyositis
Lisa G. Rider
Juvenile dermatomyositis (JDM) is a systemic autoimmune disease characterized by chronic skeletal muscle and cutaneous inflammation of unknown cause.1,2 Symptoms generally begin before age 18. JDM is relatively responsive to immunosuppressive therapy, and rapid diagnosis, and institution of adequate therapy improves outcomes.
JDM is the most common clinical subset of a larger family of disorders known as the idiopathic inflammatory myopathies (IIM) (Table 205-1).1,3,4 Juvenile polymyositis (JPM), which constitutes 2% to 8% of childhood myositis cases, has similar features without the characteristic cutaneous manifestations, but may have more severe and distal weakness. Overlap myositis, constituting 3% to 10% of childhood IIM, occurs when JDM or JPM is associated with another autoimmune disease, such as systemic lupus erythematosus, scleroderma, juvenile idiopathic arthritis, systemic vasculitis, or type I diabetes mellitus. JDM and JPM also have been reported in combination with primary immunodeficiencies such as Wiskott-Aldrich syndrome and common variable immunodeficiency, without apparent infectious triggers. Other clinical forms of IIM have been described in children, including dermatomyositis sine myositis, and focal, orbital, cancer-associated, eosinophilic, inclusion-body, and granulomatous myositis, but these subsets occur in fewer than 1 in 1 million children and are not discussed here.
The juvenile IIM also may be classified based on the presence of certain autoantibodies that are present specifically in myositis patients (myositis-specific autoantibodies), and others that are present in myositis patients as well as patients with other autoimmune diseases (myositis-associated autoantibodies). New specificities continue to be recognized. These autoantibodies tend to aid in defining patients with similar clinical and demographic features, as well as predicting immunogenetics, responses to therapy, and prognosis (Table 205-2).3,4 They are being recognized increasingly in juvenile IIM, currently with 40% to 50% of juvenile IIM patients having a defined specificity.
Table 205-1. Clinical Classification of the Juvenile Idiopathic Inflammatory Myopathies (IIM)
EPIDEMIOLOGY
The annual incidence of juvenile dermatomyositis (JDM) and juvenile polymyositis (JPM) ranges from 1.5 to 5 cases per 1 million children in various countries around the world, with an estimate from a national United States registry of new-onset JDM cases of 3.2 cases in 1 million children per year.1,5 JDM and JPM are 3.5- to 5-fold less common than adult DM and PM. JDM has a median age of onset at approximately 7 years, with a bimodal age distribution peaking at 3 to 7 years and during the early teenage years. Girls are affected 2 to 4 times more frequently than boys. The incidence is similar in different racial groups, with the overall racial distribution similar to that of the general population.
GENETICS AND PATHOPHYSIOLOGY
The pathogenesis of JDM appears to involve chronic immune activation in genetically susceptible individuals following exposure to specific environmental triggers. Evidence for genetic risk factors include an increased prevalence of other autoimmune diseases in relatives of IIM patients and rare families in which more than one relative develops an IIM.6,7 The major histocompatibility locus HLA B8-DRB1*0301-DQA1*0501 is the chief risk factor for JDM in Caucasian patients, with DQA1*0301 an additional risk factor for JDM.8-10
Group A beta-hemolytic streptococci has been associated as a risk factor in a case control study, while other organisms, including coxsackievirus B, influenza, parvovirus, and toxo-plasma, have been variably associated with JDM onset and inconsistently detected in affected muscle by polymerase chain reaction.15,19-21
Pathologically, affected muscle and cutaneous tissues demonstrate chronic perivascular and perimysial inflammation, with a predominance of plasmacytoid dendritic cells, CD4+ T cells and macrophages.1,23-25 Gene expression profiling studies have revealed a correlation between type I interferon inducible genes and disease activity in JDM and other inflammatory myopathies.26-28 Further supporting a role for these cytokines in the pathogenesis of inflammatory myopathies is the fact that IL1-α and IL-1β, which are the primary cytokines present in inflamed muscle of adult IIM patients, are produced by the infiltrating inflammatory cells as well as by ischemic muscle fibers and endothelial cells. TNF-α and transforming growth factor–β, as well as a number of other pro- and anti-inflammatory cytokines, are also variably produced by affected muscle.29
A number of promoters and inhibitors of angiogenesis are also overexpressed in the affected muscle tissue of inflammatory myositis, as are genes promoting endothelial differentiation and activation.24 In JDM, angiostatic chemokines are increased in expression and correlate with the degree of capillary loss.30 Upregulation of leucocyte adhesion molecules on the muscle arterioles and venules, particularly ICAM-1, results in the infiltration of B and CD4+ T lymphocytes, dendritic cells and macrophages.24 Proinflammatory cytokines result in damage and further infiltration of cells.
CLINICAL FEATURES AND COMPLICATIONS
JDM patients most often present with rash followed by muscle weakness.31,32 JDM is a systemic disease, with involvement of a number of organ systems (see eTable 205.1  ). Symptoms commonly develop over a period of weeks to months, although the disease may occasionally present acutely.
). Symptoms commonly develop over a period of weeks to months, although the disease may occasionally present acutely.
Muscle weakness is characteristically progressive and chronic, involving the proximal limb and axial muscles, although distal muscle weakness is evident in more severely affected children. The first symptom of JDM may be fatigue, decreased endurance, or muscle pain.33,34 A subtle decrease in activity and inability to keep up with peers in sports and play activities may be followed by progressive inability to perform daily life activities involving the proximal and axial muscles, such as climbing stairs, combing or shampooing the hair, bathing, and dressing, Ultimately, after weeks or months, the child may become unable to rise from a chair or bed. Many children exhibit the Gower sign, with difficulty rising from the floor due to hip extensor weakness, using the hands to climb up the legs as an aid. Weakness progresses at a variable and unpredictable pace in different children and not all patients reach the most severe stages.
Table 205-2. Myositis-specific and Myositis-associated Autoantibodies in Myositis
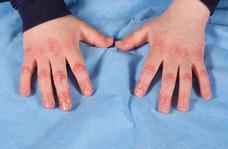
Characteristic rashes of JDM include the heliotrope (Fig. 205-1), a faint lilac to erythematous discoloration over the eyelids. This may be accompanied by periorbital edema and by Gottron papules, erythematous plaques on the extensor surfaces, particularly overlying the small joints of the hands (Fig. 205-2), but also frequently over the elbows, knees (eFig. 205.1  ), and ankles. One of these characteristic rashes is present in virtually all children with JDM.31,33 Periungual capillary changes, including dilatation, dropout, and tortuosity, are seen in over 90% of patients. These may reflect vasculopathy and active disease in other organs, particularly in the skin (eFig. 205.2
), and ankles. One of these characteristic rashes is present in virtually all children with JDM.31,33 Periungual capillary changes, including dilatation, dropout, and tortuosity, are seen in over 90% of patients. These may reflect vasculopathy and active disease in other organs, particularly in the skin (eFig. 205.2  ).35 Overgrowth of the cuticle onto the nailbed also may be seen. Malar and facial erythema, and erythematous rashes involving both sun-exposed and covered areas, are also common. Cutaneous ulceration, resulting from vasculopathy of dermal vessels, is a serious complication associated with a more severe illness course,36 but is seen in less than 30% of patients.
).35 Overgrowth of the cuticle onto the nailbed also may be seen. Malar and facial erythema, and erythematous rashes involving both sun-exposed and covered areas, are also common. Cutaneous ulceration, resulting from vasculopathy of dermal vessels, is a serious complication associated with a more severe illness course,36 but is seen in less than 30% of patients.
Dystrophic calcification of the skin, subcutaneous tissue, or muscle, often at pressure point sites, is seen in 16% to 43% of JDM patients (eFig. 205.3  ).37-39 Its development is associated with a delay in diagnosis and treatment, more severe disease, as well as cardiopulmonary involvement, and it typically becomes evident beginning 1 to 3 years after illness onset.39-40 Cellulitis, skin breakdown, joint contractures, or ulceration may develop at the sites of these deposits.40 Calcinosis may resolve over an unpredictable time frame; continued progression of calcifications, on the other hand, generally is indicative of ongoing active disease.
).37-39 Its development is associated with a delay in diagnosis and treatment, more severe disease, as well as cardiopulmonary involvement, and it typically becomes evident beginning 1 to 3 years after illness onset.39-40 Cellulitis, skin breakdown, joint contractures, or ulceration may develop at the sites of these deposits.40 Calcinosis may resolve over an unpredictable time frame; continued progression of calcifications, on the other hand, generally is indicative of ongoing active disease.
Involvement of the striated and smooth muscle of the gastrointestinal tract, resulting in difficulty swallowing or handling secretions, is a sign of severe disease requiring aggressive treatment. Subclinical abnormalities on modified barium swallow examination may be seen in up to 80% of JDM patients.41 Such patients are at risk for aspiration pneumonia. Vasculitis of the gastrointestinal tract, observed in less than 10% of JDM patients, may result in severe abdominal pain, gastrointestinal bleeding, or perforation.42 Abdominal pain that is persistent, progressive or severe should be carefully evaluated, and stool should be tested for occult blood. Children not at imminent risk of perforation may undergo barium examination or contrast-enhanced computed tomography, which may demonstrate dilation or thickening of the bowel wall, intra-luminal air or evidence of bowel necrosis.43 Lower gastrointestinal tract dysmotility, resulting in constipation, abdominal pain, or bloating, also may occur as a result of smooth muscle involvement.
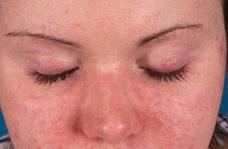
FIGURE 205-1. Heliotrope rash, a purplish discoloration over the eyelids with associated periorbital edema, which is characteristic of dermatomyositis. This child also has a malar rash.
Palatal involvement leading to hoarseness and dysphonia is seen in up to 45% of patients.31,33 Respiratory muscle weakness with dyspnea on exertion and restrictive lung disease is common,44 but interstitial lung disease is relatively infrequent in juvenile IIM. Arthritis is present in two thirds of patients, and typically involves the knees, wrists, elbows, or interphalangeal joints.45 Contractures of large joints often accompany severe, persistent muscle weakness or may be seen in isolation. Tenosynovitis also may be present. Constitutional symptoms, including fatigue, fever, Raynaud phenomenon, and lymphadenopathy, may be noted, particularly at the onset of illness.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


