Fig. 45.1
a and b Intraoperative photographs showing prolapsed rectal polyps
Colo-colonic intussusception is unusual and rarely leads to prolapse of the intussusceptum.
Juvenile polyps are variable in size, with clinical symptoms developing from polyps that range in size from < 0.5 cm up to ≥ 3 cm.
Most juvenile polyps are pedunculated and bleed easily (Fig. 45.2).
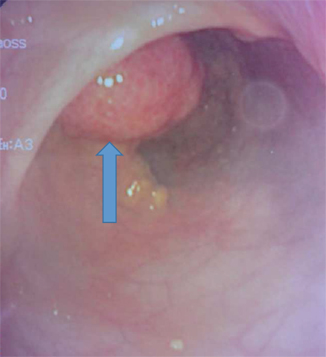
Fig. 45.2
Intraoperative colonoscopic photograph showing a large colonic polyp diagnosed and removed via colonoscopy
They are mucosal lesions that contain dilated, mucous-filled glands with inflammation in the lamina propria; on cut surface, they often appear cystic. This combination of findings leads to the common reference to these as inflammatory or retention polyps.
Juvenile polyps are hamartomas.
In contrast to juvenile polyposis syndrome, juvenile polyps are not neoplastic and are not associated with malignant transformation. There have been reports of adenomatous changes in juvenile polyps and though rare, dysplasia may confer an increased risk of carcinoma.
Many of these polyps simply outgrow their blood supply, become ischemic, and autoamputate.
Juvenile polyps are usually confined to the rectosigmoid colon. However, between 50 and 60 % of patients have more than one polyp.
About 25 % of patients have polyps in the cecum or ascending colon.
Patients with an isolated episode of bleeding that is self-limited and who pass tissue in the stool should be observed.
Patients with persistent or recurrent bleeding, or who are having pain or other symptoms should have a colonoscopy (Fig. 45.3).
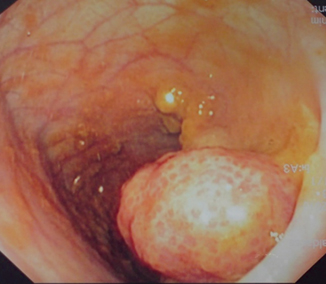
Fig. 45.3
Intraoperative colonoscopic photograph showing a large juvenile polyp which bleeds easily. This was removed endoscopically
When the diagnosis is made with colonoscopy, a polypectomy can safely be performed.
The recent evidence that patients can have more than one polyp and that polyps may be found in the cecum and ascending colon makes colonoscopy more preferable to the traditional recommendation of proctoscopy or flexible sigmoidoscopy.
Familial Juvenile Polyposis Syndrome
Juvenile polyposis syndrome is a rare polyposis syndrome that runs in families.
Compared to children having a single juvenile polyp, those with Juvenile polyposis syndrome have multiple polyps.
The exact number of juvenile polyps necessary to diagnose a polyposis syndrome is controversial.
Patients with familial juvenile polyposis are diagnosed when the following criteria are met:
More than five (some say ten) juvenile polyps in the colon
Multiple juvenile polyps throughout the GI tract
Any number of juvenile polyps with a family history of juvenile polyposis
Although most polyps develop in the colon and small bowel, they can also occur in the stomach.
These polyps are hamartomas and the term “juvenile” refers to the type of polyps seen in these patients, not the age of onset.
Familial juvenile polyposis is an autosomal dominant disorder with varied presentation. About 75 % of patients will have a parent with the syndrome, but 25 % do not have a family history suggesting a new mutation.
Mutations in the BMPR1A or SMAD4 gene have been identified, but together these account for only 40 % of patients.
Children with first-degree relatives who have juvenile polyposis syndrome should undergo screening colonoscopy starting at age 12 years.
Associated congenital extra colonic anomalies are found in as many as 20 % of these patients including:
Macrocephaly, hydrocephalus, and spina bifida
Congenital heart disease
Urogenital anomalies including undescended testes, bifid uterus and vagina, abnormal pelvi-ureteric junction insertion, unilateral renal agenesis
Osteoma
Malrotation, Meckel diverticulum
Familial juvenile polyposes affect 1:100,000 live births.
Familial juvenile polyposes have a cumulative risk of malignancy > 50 %.
Surgical treatment options:
Endoscopic removal of the colon polyps followed by yearly endoscopy with surveillance biopsies.
Laparotomy, enterotomies, and polypectomies.
If there are clusters of polyps in isolated areas, limited segmental resection may be appropriate.
Patients with numerous polyps in the colon will require proctocolectomy with ileoanal anastomosis.
Prophylactic colectomy with ileorectal anastomosis is recommended for children with familial juvenile polyposis who have:
Severe or repeated bleeding
Hypoprotinemia
Failure to thrive
Peutz–Jeghers Syndrome
The broad category of hamartomatous polyposis syndromes encompasses several syndromes, mainly:
Peutz–Jeghers syndrome (PJS)
Phosphatase and tensin homolog (PTEN)-associated hamartomatous syndromes including:
◦ Cowden syndrome
◦ Bannayan–Riley–Ruvalcaba syndrome (BRR)
Familial juvenile polyposis syndrome
Cronkhite–Canada syndrome
The association of intestinal polyposis with mucocutaneous pigmentation was first reported in three generations of a Dutch family by Peutz in 1921. In 1949, Jeghers reported ten similar patients establishing the now well-known syndrome.
In PJS:
There are hamartomatous polyps which can occur anywhere within the GI, most commonly in the jejunum.
There are characteristic melanin spots on the lips, around the mouth, on the inside of cheeks and digits.
Patients with Peutz–Jeghers syndrome have multiple pedunculated hamartomatous polyps distributed as follows:
The small intestine (78 %)
Colon (42 %)
Stomach (38 %)
Rectum (28 %)
PJS has an estimated prevalence of between 1:120,000 and 1:200,000 live births.
These polyps are often large and may cause intussusception with intestinal obstruction.
Polyps within ureter, bladder, and renal pelvis have also been reported in patients with PJS as well as nasal and bronchial polyps.
PJS often runs in families but may occur without any known family member affected.
These children often require multiple surgeries due to recurrent intussusception.
PJS has been localized via gene linkage and logarithm of odds (LOD) score to mutations in band 19p13.3–13.4, which is now known to encode a serine-threonine kinase (STK11/LKB1) within this region.
Approximately 80 % of patients with PJS have this gene mutation.
PJS is inherited as an autosomal dominant.
Surgical management:
Most episodes of abdominal pain in patients with the Peutz–Jeghers syndrome which are usually due to intussusceptions are self-limited and resolve without surgical intervention.
The main indication for operative treatment is the presence of intestinal obstruction.
If an operation is not delayed too long, the intussusception can be reduced followed by an enterotomy and polypectomy without segmental small bowel resection.
The entire GI should be palpated and any other polyps removed.
If the patient presents late and the intussusception cannot be reduced, resection with primary anastomosis should be performed.
Patients with Peutz–Jeghers syndrome are at increased risk of:
Gastrointestinal malignancies of:
◦ Small intestines
◦ Colon
◦ Pancreatico-biliary
Extraintestinal malignancy of the:
◦ Breast
◦ Ovary
◦ Cervix
◦ Germ cell tumors
Development of gynecomastia commonly precedes the development of gynecologic or testicular malignancy.
It is estimated that the risk of cancer is increased 18-fold over the general population.
None of these cancers occurs at a frequency that justifies prophylactic removal of the organs, but awareness of the risk should guide surveillance.
Familial Adenomatous Polyposis
FAP is the most common intestinal polyposis syndrome with an estimated frequency of 1:13,000 births .
It is inherited as an autosomal dominant disorder.
FAP runs in families but may occur sporadically.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


