Inherited Bleeding Disorders
Amy L. Dunn, Shannon L. Meeks, and Thomas C. Abshire
Inherited bleeding disorders are a heterogeneous group of conditions that lead to excessive hemorrhage. The disorders have distinct inheritance patterns, clinical bleeding symptoms, and laboratory abnormalities. Therefore, the history and physical examination are key to guiding appropriate laboratory testing and diagnosis.
THE HEMOPHILIAS
Hemophilia A, or classical hemophilia, results from congenital deficiency or absence of circulating factor VIII (FVIII). It is an X-linked recessive disorder with an incidence of approximately 1:5000 male births.1 Hemophilia B is also known as Christmas disease.2 It results from a congenital deficiency or absence of factor IX (FIX). It is an X-linked recessive disorder with an incidence of approximately 1:25,000 male births.1 Hemophilia C is an autosomal recessive disorder resulting in decreased amounts of circulating factor XI. It affects 1 in 100,000 people.
 FACTOR STRUCTURE AND FUNCTION
FACTOR STRUCTURE AND FUNCTION
Factor VIII is a plasma glycoprotein.3 The encoding gene is found on the long arm of the X chromosome (Xq28). The majority of FVIII is thought to be synthesized in liver endothelial cells. Upon release into the circulation, it is immediately noncovalently linked to von Willebrand factor (vWF). This prevents enzymatic degradation of FVIII until it is needed during coagulation. 
Factor IX is a vitamin K–dependent serine protease believed to be synthesized in the liver and released into the circulation in its inactive form. The gene is on the end of the X chromosome (Xq27).3 During coagulation, tissue factor and FVII activate FIX. Factor IXa in turn activates FVIII. 
FXI is a serine protease that participates in the contact pathway.5 It is synthesized in the liver and circulates in its inactive form as a homodimer. The gene encoding FXI is on chromosome 4q32-35. Its major role appears to be activation of FIX. Bleeding in hemophilia C is likely related to inadequate FIX activation or lack of production of thrombin-activatable fibrinolysis inhibitor.6
 CLINICAL MANIFESTATIONS
CLINICAL MANIFESTATIONS
The hallmark of hemophilia A– and B–related bleeding is delayed bleeding, along with joint and muscle hemorrhage. In comparison, patients with von Willebrand disease and hemophilia C more commonly manifest mucocutaneous bleeding. As hemophilia A and B are X-linked, the vast majority of affected patients are male. Females can be affected in cases of extreme X chromosome lyonization or gene abnormalities such as Turner syndrome. 
In general, the severity of bleeding in hemophilia A and B depends upon the percentage of circulating clotting factor activity. Patients with levels of greater than 5% to 40% are classified as having mild hemophilia, those with levels of 1% to 5% as moderate, and those with less than 1% activity as having severe disease. Commonly, patients with severe disease will suffer from spontaneous bleeding, and those with mild-moderate disease more typically bleed with trauma or surgery. In the newborn period, the most common symptoms are bleeding in the brain and at sites of blood withdrawal, immunizations, and circumcision. Older children and adults may experience excessive bruising, hematomas, and intracranial, joint, muscle, and mouth bleeding.
Patients with hemophilia C have a heterogeneous bleeding pattern that is not directly related to circulating levels.5 In general, patients with less than 20% activity are considered to have severe disease. It is rare, however, for patients, even those with low levels, to suffer from spontaneous bleeding.
In general, patients with less than 20% activity are considered to have severe disease. It is rare, however, for patients, even those with low levels, to suffer from spontaneous bleeding.
 LABORATORY FINDINGS
LABORATORY FINDINGS
An elevated active partial thromboplastin time and decreased plasma FVIII, IX, or XI activity assay confirm the diagnosis. 
 TREATMENT
TREATMENT
The mainstay of hemophilia A and B care is intravenously delivered factor concentrates. Factor VIII and IX concentrates can be purified from plasma or recombinantly synthesized.7,8 The plasma-derived FVIII products contain varying amounts of vWF, depending upon the manufacturing process. Due to a history of viral contamination of plasma pools, both types of products now undergo multiple viral and pathogen attenuation steps. Importantly, no infectious complications have been reported since these steps were incorporated into the manufacturing process. 
Factor infusions can be delivered either in response to bleeding episodes (demand therapy) or to prevent bleeding (prophylaxis). To prevent or minimize long-term sequelae, demand therapy should be given as soon as possible after a bleeding episode is recognized. 
In developed countries, prophylactic therapy delivered 1 to 4 times per week dosed to keep trough levels above 1% is considered the standard of care. If prophylactic infusions are started at a young age prior to the development of arthropathy, it is termed primary prophylaxis and is the only therapy proven to prevent the long-term complication of degenerative joint disease.10 It is common practice to begin prophylaxis prior to the onset of recurrent joint bleeding.
It is common practice to begin prophylaxis prior to the onset of recurrent joint bleeding.
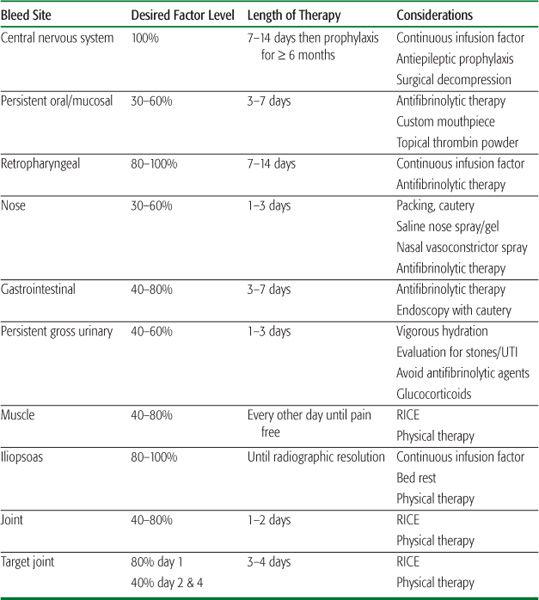
The dose and frequency of factor delivery is calculated based upon the half-life of the product (typically 10 to 12 hours for FVIII and 20 to 24 hours for FIX), the intravascular volume of distribution (1 international unit [IU] of FVIII per kilogram raises the plasma concentration by about 2%, and 1 IU of FIX per kilogram raises the plasma concentration by about 1%), and the desired clotting factor activity. Forty percent activity is considered hemostatic in most cases; however, in the setting of surgery or life- or limb-threatening hemorrhage, higher levels are necessary.  Table 436-1 illustrates an approach to factor replacement therapy of commonly encountered bleeding events.14
Table 436-1 illustrates an approach to factor replacement therapy of commonly encountered bleeding events.14
Desmopressin or DDAVP (1-deamino-8-D-argine vasopressin) is a synthetic form of the hormone vasopressin. DDAVP causes release of FVIII and vWF from storage sites along the endothelium and within platelet storage granules.15 Patients with mild or moderate hemophilia A can be tested with this product, and their response (rise in FVIII level) can be measured. If patients show a response by manifesting hemostatic levels of FVIII, this product is often sufficient to treat or prevent bleeding. FVIII storage pools become depleted after multiple doses, so this treatment is not adequate for lengthy therapy. Additionally, fluid intake must be monitored closely as severe hyponatremia may result.16 Antifibrinolytic therapy to stabilize the fibrin clot is particularly useful in diminishing bleeding symptoms in locations with prominent fibrinolytic activity, such as the mouth, gastrointestinal tract, and uterus.
Antifibrinolytic therapy to stabilize the fibrin clot is particularly useful in diminishing bleeding symptoms in locations with prominent fibrinolytic activity, such as the mouth, gastrointestinal tract, and uterus.
Therapy for patients with hemophilia C relies upon replacement of FXI with fresh-frozen plasma, the use of antifibrinolytic agents, and fibrin glue.5
 COMPLICATIONS
COMPLICATIONS
Inhibitor Development
A common complication of hemophilia is development of inhibitory antibodies to the infused factor. These inhibitory antibodies occur in approximately 30% of patients with severe hemophilia A and to a lesser extent in mild and moderate disease.19 They are more likely to occur in the setting of a positive family history of inhibitors in association with large gene deletions such as introns 1 and 22 inversions and in patients who are not of European ancestry. They often develop within the first 20 exposures to exogenous FVIII, but can occur at any age. The antibody titer is measured using a Bethesda assay and is expressed in Bethesda units (BU).25 Low titer inhibitors less than 5 BU are often transient and of little clinical significance, whereas those 5 BU or greater can significantly impact patient care and quality of life. Immune tolerance therapy with repeated exposure to FVIII concentrate over a period of months to years may eradicate the antibody. Bleeding in the setting of a high titer inhibitor often requires bypassing therapy with either high doses of recombinant FVIIa or a prothrombin complex concentrate. 
Table 436-1. Suggested Approach to Treatment of Bleeding Episodes in Hemophilia
Hemophilia B and C are rarely complicated by the development of inhibitory antibodies. While these antibodies occur in only approximately 3% of patients with severe hemophilia B, anaphylactoid reactions to exogenous FIX that in some cases are life-threatening have been reported prior to recognition of a positive antibody titer.27 Successful desensitization to FIX has been achieved in some patients, and immune tolerance therapy with repeated exposure to FIX concentrate may eradicate the antibody.28 Nephrotic syndrome, however, is a uniquely reported complication of immune tolerance therapy with FIX products and commonly does not occur until several months into therapy. 
Hemophilic Arthropathy
Degenerative joint disease due to recurrent hemarthrosis is the single largest preventable cause of morbidity for patients with hemophilia A and B. Recurrent spontaneous or trauma-induced joint bleeding is often seen in children and adults with severe hemophilia. Although the pathogenesis of the resultant arthropathy is multifactorial, iron is the most likely trigger of the degenerative changes. 
VON WILLEBRAND DISEASE
von Willebrand disease (vWD) was first described in 1926 by Erik von Willebrand in a group of patients with an autosomal inherited bleeding pattern. It is characterized by deficiency of vWF and is the most common inherited bleeding disorder, approaching an incidence of 0.1% to 1%. Two major functions of the vWF protein are to mediate adhesion of platelets to sites of vascular injury via interaction with platelet glycoprotein Ib and to the subendothelial matrix (collagen). It also serves as a carrier protein for FVIII. vWF can only mediate platelet adhesion normally if it is assembled into large multimers. Accordingly, defects in vWF may cause bleeding by impairing both platelet adhesion and (more rarely) blood clotting. The clinical manifestations vary but usually comprise mucosal bleeding symptoms (bruising, nose bleeding, mouth/teeth-related bleeding, and menorrhagia) and bleeding immediately after invasive procedures or surgery. Von Willebrand disease is thought to be caused by an inherited mutation in the vWF gene located on chromosome 12p, but there may also be certain modifying factors and/or genes, especially in type 1 von Willebrand disease, that modulate its expression (eFig. 436.3  ). The disorder typically demonstrates an autosomal dominant inheritance pattern, although several subtypes are inherited in a recessive or doubly heterozygous fashion.50 The classification for von Willebrand disease (Fig. 436-1) includes 3 major categories: partial quantitative deficiency (type 1), qualitative deficiency (type 2), and total deficiency (type 3). Qualitative abnormalities (type 2) are further divided into four variants or subtypes (2A, 2B, 2M, and 2N).
). The disorder typically demonstrates an autosomal dominant inheritance pattern, although several subtypes are inherited in a recessive or doubly heterozygous fashion.50 The classification for von Willebrand disease (Fig. 436-1) includes 3 major categories: partial quantitative deficiency (type 1), qualitative deficiency (type 2), and total deficiency (type 3). Qualitative abnormalities (type 2) are further divided into four variants or subtypes (2A, 2B, 2M, and 2N).
 DIAGNOSIS
DIAGNOSIS
The clinical history is the most important criterion used in establishing a diagnosis of von Willebrand disease.51 Without a history of significant cutaneous or mucosal bleeding in the patient or a family history of bleeding, it is difficult to make a diagnosis. However, children as a group often have not been carefully questioned nor have they been hemostatically stressed; therefore, an underlying bleeding disorder might be missed. Laboratory studies are directed at documenting von Willebrand factor (vWF) deficiency or qualitative defects in vWF. Screening tests (prothrombin time, activated partial thromboplastin time, fibrinogen, thrombin time, and platelet function analyzer) are often normal, so specific testing for von Willebrand disease must be performed. The von Willebrand disease screening tests include von Willebrand factor antigen, ristocetin cofactor, FVIII activity, and vWF multimers tests. Abnormal values for von Willebrand factor antigen, ristocetin cofactor, and FVIII activity are usually less than 0.5 IU/dL (50%).52 There are a number of modulating factors that are associated with lower vWF levels, such as blood type O and hypothyroidism.53 Accordingly, for levels of 30% to 50%, caution must be exercised in rendering a diagnosis, particularly if the bleeding symptoms are minimal. Certain physiologic conditions such as pregnancy, the menstrual cycle, exercise and stress (blood drawing, bleeding, or acute infection) may elevate the vWF level. Conversely, if the patient has definite bleeding symptoms and levels are above 50%, these results may have been confounded by some of the previously described variables, and testing should be repeated in a carefully controlled setting. 
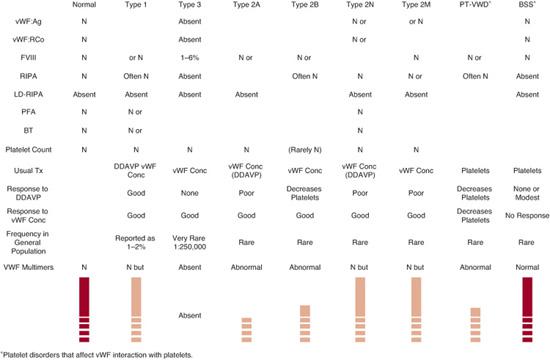
FIGURE 436-1. Summary of von Willebrand disease (vWD) types, types 1, 3, 2A, 2B, 2M, 2N) and absence or defective platelet glycoprotein Ib receptor for vWF, platelet-type vWD (PT-vWF), or Bernard Soulier Syndrome (BSS). The lower portion illustrates the vWF multimers that are identified by sodium dodecylsulfate-agarose gel electrophoresis of vWF for each of these variants. BT, bleeding time; Conc, concentrate; LD-RIPA, ristocetin-induced platelet aggregation to low-dose ristocetin; N, normal; PFA, platelet function analyzer; Tx, treatment; vWF:Ag, von Willebrand factor antigen; vWF:RCo, vWF activity by ristocetin cofactor assay. (Source: Courtesy of RR Montgomery.)
Type 1 von Willebrand Disease
Type 1 von Willebrand disease accounts for more than 75% to 80% of patients with von Willebrand disease and is caused by partial, quantitative vWF deficiency with proportional reduction in all the von Willebrand disease screening tests.54 Unlike patients with type 2 disease, many patients with type 1 von Willebrand disease do not have a defined gene mutation. Neonates with type 1 disease rarely bleed, and establishment of the diagnosis is difficult due to high levels of vWF in the fetus and newborn. It is important to recognize a form of type 1 von Willebrand disease that has increased clearance (half-life < 1–2 hours) of the vWF (von Willebrand disease Vicenza).55
Type 2 von Willebrand Disease
A qualitative abnormality of the von Willebrand factor (vWF) protein with a disproportionate reduction of ristocetin cofactor compared to von Willebrand factor antigen is the hallmark of the type 2 von Willebrand disease defect, which accounts for 15% to 20% of patients with von Willebrand disease. Often, there is a defective vWF multimeric pattern. Most type 2 von Willebrand disease variants have known genetic mutations.56
Type 2A von Willebrand Disease On laboratory testing, a disproportionately low ristocetin cofactor relative to von Willebrand factor antigen reflects the decreased affinity of vWF for platelets due to an absence of large vWF multimers. Type 2A von Willebrand disease is transmitted as a dominant disorder. The loss of large and intermediate-sized vWF multimer forms is due to either impaired protein production or by proteolysis from the ADAMTS-13 metalloprotease. This type accounts for 10% to 12% of von Willebrand disease cases.
Type 2B von Willebrand Disease Type 2B von Willebrand disease occurs in 3% to 5% of von Willebrand disease subjects and is characterized by increased affinity of the abnormal vWF for the platelet glycoprotein Ib, followed by clearance of both the higher-molecular-weight multimers of vWF and platelets. The ristocetin cofactor is not as reduced as compared with von Willebrand factor antigen as seen in type 2A von Willebrand disease. The thrombocytopenia seen in type 2B disease may be intermittent and often is exacerbated by stress, infection, or pregnancy. Type 2B von Willebrand disease may initially be misdiag-nosed as ITP or a more rare type of von Willebrand disease known as pseudo or platelet-type von Willebrand disease. In platelet-type von Willebrand disease, the defect is in the platelet (glycoprotein Ib receptor) and not in the vWF. The diagnosis of type 2B von Willebrand disease depends upon aggregation to low doses of ristocetin used in platelet aggregation testing.
Type 2M von Willebrand Disease von Willebrand disease type 2M occurs in 1% to 2% of the von Willebrand disease population and is due to decreased affinity to glycoprotein Ib, the opposite of type 2B von Willebrand disease. Laboratory results reveal a disproportionate decrease (approximately 50%) of ristocetin cofactor compared with von Willebrand factor antigen and a normal vWF multimer pattern.
Type 2N von Willebrand Disease Mutations that selectively inactivate the FVIII-binding site on vWF produce an autosomal recessive von Willebrand disease phenotype in which the platelet-dependent functions of vWF are preserved, but FVIII levels are low (< 10%). This defect occurs in 1% to 2% of the von Willebrand disease population and is known as type 2 Normandy. These patients may be misdiagnosed as having hemophilia A, although the inheritance pattern suggests an autosomal pattern. Test results typically show near-normal vWF levels and a prolonged activated partial thromboplastin time secondary to the low FVIII. Definitive diagnosis is made by demonstrating lack of effect of recombinant FVIII infusion, FVIII-binding studies, and/or molecular studies that confirm the type 2N mutation in the vWF gene.
Type 3 von Willebrand Disease
This type of von Willebrand disease is characterized by the complete absence of vWF, with no functional activity and no antigenic protein. It is inherited as a recessive disorder. FVIII is in the moderate hemophilia range (3–5% FVIII activity). This type of von Willebrand disease is extremely rare  .
.
 TREATMENT OF VON WILLEBRAND DISEASE
TREATMENT OF VON WILLEBRAND DISEASE
For minor bleeding (nose and mouth related), DDAVP and/or antifibrinolytic therapy are recommended.57 For major bleeding or surgery, intravenous DDAVP or FVIII/vWF-containing concentrates are used. Repeated doses of DDAVP may result in tachyphylaxis; therefore, dosing is generally given every 24 to 48 hours as an inpatient and usually for only 3 days in the outpatient setting. Facial flushing is a common side effect. Blood pressure and serum sodium should be monitored, and patients should be fluid restricted to slightly below maintenance levels to avoid hyponatremia and possible seizures. This complication is most frequent in infants or in patients receiving excessive intravenous fluids.
For major bleeding or surgery, intravenous DDAVP or FVIII/vWF-containing concentrates are used. Repeated doses of DDAVP may result in tachyphylaxis; therefore, dosing is generally given every 24 to 48 hours as an inpatient and usually for only 3 days in the outpatient setting. Facial flushing is a common side effect. Blood pressure and serum sodium should be monitored, and patients should be fluid restricted to slightly below maintenance levels to avoid hyponatremia and possible seizures. This complication is most frequent in infants or in patients receiving excessive intravenous fluids.
Type 1 von Willebrand disease patients with an inadequate response to DDAVP, most patients with type 2 von Willebrand disease variants, and those with type 3 von Willebrand disease must be treated with virally inactivated FVIII/vWF-containing concentrates. Some patients with types 2A and 2M disease will have a favorable response to DDAVP, but the duration of response is often shorter than in patients with type 1 von Willebrand disease. In patients with type 2B von Willebrand disease, DDAVP releases the abnormal vWF from endothelial cells and may worsen the thrombocytopenia. Platelet-type von Willebrand disease should be treated with platelet concentrates. Transfusion with fresh-frozen plasma or cryoprecipitate should no longer be used because these products (particularly fresh-frozen plasma) do not contain enough vWF and cannot be virally inactivated (cryoprecipitate).
DEFECTS IN THE TISSUE FACTOR (EXTRINSIC) PATHWAY
Factor VII is a 50-kDa, vitamin K–dependent serine protease that is produced in the liver. Deficiency is rare, with a prevalence of 1:500,000. Homozygotes can have a severe bleeding phenotype similar to classical hemophilia, whereas heterozygotes are typically asymptomatic. In the majority of patients, FVII deficiency is associated with a mild bleeding phenotype. Isolated prolongation of the prothrombin time is found in the initial workup and is confirmed with a low FVII activity level. First-line treatment for bleeding in FVII deficiency is recombinant FVIIa. Antifibrinolytic agents may be beneficial for mucosal bleeding. 
DEFECTS IN THE COMMON PATHWAY 
Factors II, V, and X are essential parts of the common pathway of coagulation, and deficiencies in any of these factors can lead to prolongation of the prothrombin time and activated partial thromboplastin time. 
Homozygous or compound heterozygous defects in the factor II, V, or X genes can lead to moderate to severe bleeding symptoms, with patients having FX deficiency more likely to manifest severe symptoms. In general, lower levels of protein are associated with more severe bleeding. Reported bleeding symptoms include easy bruising, mucosal bleeding, surgical bleeding, and trauma-related bleeding, whereas hemarthroses and intracranial hemorrhage are less common. Acute bleeding episodes can be treated with fresh-frozen plasma. For FII or X deficiency, another treatment option is a prothrombin complex concentrate. Platelets can also be given for FV deficiency. Antifibrinolytic therapies may be administered to treat mucosal bleeding. 
DYSFIBRINOGINEMIA AND AFIBRINOGINEMIA
Fibrinogen is a 340-kDa glycoprotein that is primarily synthesized in the liver. It has a hemostatic level of approximately 50 mg/dL. When initiated by a hemostatic challenge, the coagulation cascade produces thrombin that then cleaves fibrinogen to fibrin, allowing polymerization and formation of a fibrin clot. Mutations in the fibrinogen genes may be inherited in an autosomal recessive or an autosomal dominant fashion. Homozygotes or compound heterozygotes for severe mutations have afibrinogenemia in which no fibrinogen is produced. Patients with hypofibrinogenemia have a decreased amount of a normal fibrinogen protein. Dysfibrinogenemia typically results from mutations that alter the structure of the fibrinogen protein leading to different functional properties. The incidence of afibrinogenemia is approximately 1 in 1 million people. The incidence of dysfibrinogenemia is unknown.58,59,63,65 Patients with afibrinogenemia have an abnormal prothrombin time, activated partial thromboplastin time, and bleeding time. Prolonged thrombin and reptilase clotting times are seen in dysfibrinogenemias, and immunoassays showing discrepant protein activity and antigen levels confirm the diagnosis. 
 CLINICAL FEATURES
CLINICAL FEATURES
Manifestations in patients with afibrinogenemia commonly include umbilical cord and mucosal bleeding, but joint and muscle bleeding, intracranial hemorrhage, and bleeding with surgery or trauma occur as well. Patients with hypofibrinogenemia typically have a less severe bleeding phenotype. Women with afibrinogenemia and hypofibrinogenemia have an increased risk of miscarriage. Splenic rupture has been reported in patients with afibrinogenemia.63,66 Patients with dysfibrinogenemia have a diverse clinical phenotype, ranging from asymptomatic to severe bleeding, delayed wound healing, and/or thrombosis. 
Cryoprecipitate is commonly used for treatment and prophylaxis. For mucosal bleeding, treatment with an antifibrinolytic agent is helpful. Antibody development to fibrinogen replacement therapy is rare, but anaphylaxis has been reported. 
FACTOR XIII DEFICIENCY
FXIII deficiency is an autosomal recessive disorder with a prevalence of approximately 1 in 2 million people. When activated, FXIII cross-links the fibrin clot, which is then resistant to fibrinolysis. Most patients present early in life with severe or life-threatening bleeding. Bleeding from the umbilicus shortly after birth is seen in 80% of untreated patients, and 30% of patients have intracranial hemorrhage.67 Excessive bruising, muscle bleeding, joint bleeding, menorrhagia, and bleeding following surgery or trauma also occur. Recurrent miscarriage is seen unless treatment is given during pregnancy. There are also reports of delayed wound healing. The routine screening tests for hemostasis are all normal. As a qualitative screening test, clot solubility is increased. Confirmatory testing with FXIII activity levels can be performed.58,59,63,68 Patients with FXIII deficiency can be treated with FXIII concentrates as well as fresh-frozen plasma and cryoprecipitate. 
DEFECTS IN THE FIBRINOLYTIC PATHWAY
In the fibrinolytic system, plasminogen is activated primarily by tissue plasminogen activator to plasmin, which in turns breaks down both fibrin and fibrinogen. The fibrinolytic system is regulated by plasminogen activator inhibitor-1, which inhibits the activation of plasminogen, and by αα2-antiplasmin, which inhibits plasmin. Autosomally inherited deficiencies in plasminogen activator inhibitor 1 and αα2-antiplasmin result in unopposed fibrinolysis by plasmin and lead to ineffective hemostasis and bleeding. Bleeding symptoms range from moderate to severe and include soft tissue hematomas, mucocutaneous bleeding, hemarthroses, and bleeding following surgery or trauma. All of the routine coagulation tests, including the prothrombin time, partial thromboplastin time, bleeding time, and thrombin time, are normal. Treatment with anti-fibrinolytic agents prevents the binding of plasminogen to fibrin, thereby inhibiting fibrinolysis and stabilizing the fibrin clot. 
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


