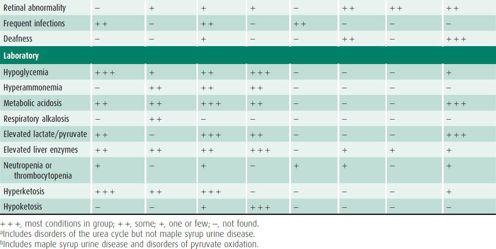
Table 36–2. Obtaining and handling samples to diagnose inborn errors.

Laboratory studies are almost always needed for the diagnosis of inborn errors. Serum electrolytes and pH should be used to estimate anion gap and acid-base status. Serum lactate, pyruvate, and ammonia levels are available in most hospitals, but care is needed in obtaining samples appropriately. Amino acid, acylcarnitine, and organic acid studies must be performed at specialized facilities to ensure accurate analysis and interpretation. An increasing number of inborn errors are diagnosed with DNA sequencing, but interpretation of private mutations, that is, mutations only seen in a particular family, can be problematic. Knowing the causative mutation in the family allows prenatal diagnosis to be done by molecular analysis. This can be done on any material that contains fetal DNA such as chorionic villi, amniotic cells, or fetal blood obtained through umibilical cord blood sampling.
The physician should know what conditions a test can detect and when it can detect them. For example, urine organic acids may be normal in patients with medium-chain acyl-CoA dehydrogenase deficiency or biotinidase deficiency; glycine may be elevated only in cerebrospinal fluid (CSF) in patients with glycine encephalopathy. A result that is normal in one physiologic state may be abnormal in another. For instance, the urine of a child who becomes hypoglycemic upon prolonged fasting should be positive for ketones. In such a child, the absence of ketones in the urine suggests a defect in fatty acid oxidation.
Samples used to diagnose metabolic disease may be obtained at autopsy. Samples must be obtained in a timely fashion and may be analyzed directly or stored frozen until a particular analysis is justified by the results of postmortem examination, new clinical information, or developments in the field. Studies of other family members may help establish the diagnosis of a deceased patient. It may be possible to demonstrate that parents are heterozygous carriers of a particular disorder or that a sibling has the condition.
COMMON CLINICAL SITUATIONS
1. Mental Retardation
Some inborn errors can cause mental retardation without other distinguishing characteristics. Measurements of serum amino acids, urine organic acids, and serum uric acid should be obtained in every patient with nonspecific mental retardation. Urine screens for mucopolysaccharides and succinylpurines, and serum testing for carbohydrate-deficient glycoproteins are useful because these disorders do not always have specific physical findings. Absent speech can point to disorders of creatine. Abnormalities of the brain detected by magnetic resonance imaging can suggest specific groups of disorders (eg, cortical migrational abnormalities in peroxisomal biogenesis disorders).
2. Acute Presentation in the Neonate
Acute metabolic disease in the neonate is most often a result of disorders of protein or carbohydrate metabolism and may be clinically indistinguishable from sepsis. Prominent symptoms include poor feeding, vomiting, altered mental status or muscle tone, jitteriness, seizures, and jaundice. Acidosis, alkalosis, or altered mental status out of proportion to systemic symptoms should increase suspicion of a metabolic disorder. Laboratory measurements should include electrolytes, ammonia, lactate, glucose, blood pH, and urine ketones and reducing substances. Amino acids in CSF should be measured if glycine encephalopathy is suspected. Serum and urine amino acid, urine organic acid, and serum acylcarnitine analysis should be performed urgently. Neonatal cardiomyopathy or ventricular arrhythmias should be investigated with serum acylcarnitine analysis.
3. Vomiting & Encephalopathy in the Infant or Older Child
Electrolytes, ammonia, glucose, urine pH, urine reducing substances, and urine ketones should be measured in all patients with vomiting and encephalopathy before any treatment affects the results. Samples for serum amino acids, serum acylcarnitine profile, and urine organic acid analysis should be obtained early. In the presentation of a Reye-like syndrome (ie, vomiting, encephalopathy, and hepatomegaly), amino acids, acylcarnitines, carnitine levels, and organic acids should be assessed immediately. Hypoglycemia with inappropriately low urine or serum ketones suggests the diagnosis of fatty acid oxidation defects.
4. Hypoglycemia
Duration of fasting, presence or absence of hepatomegaly, and Kussmaul breathing provide clues to the differential diagnosis of hypoglycemia. Serum insulin, cortisol, and growth hormone should be obtained on presentation. Urine ketones, urine organic acids, plasma lactate, serum acylcarnitine profile, carnitine levels, ammonia, triglycerides, and uric acid should be measured. Ketone body production is usually not efficient in the neonate, and ketonuria in a hypoglycemic or acidotic neonate suggests an inborn error. In the older child, inappropriately low urine ketone levels suggest an inborn error of fatty acid oxidation. Assessment of ketone generation requires simultaneous measurements of quantitative serum 3-hydroxybutyrate, acetoacetate, and free fatty acids in relation to a sufficient duration of fasting and age. Metabolites obtained during the acute episode can be very helpful and avoid the need for a formal fasting test.
5. Hyperammonemia
Symptoms of hyperammonemia may appear and progress rapidly or insidiously. Decreased appetite, irritability, and behavioral changes appear first with vomiting, ataxia, lethargy, seizures, and coma appearing as ammonia levels increase. Tachypnea inducing respiratory alkalosis due to a direct effect on respiratory drive is characteristic. Physical examination cannot exclude the presence of hyperammonemia, and serum ammonia should be measured whenever hyperammonemia is possible. Severe hyperammonemia may be due to urea cycle disorders, organic acidemias, or fatty acid oxidation disorders (such as carnitine-acylcarnitine translocase deficiency) or, in the premature infant, transient hyperammonemia of the newborn. The cause can usually be ascertained by measuring quantitative serum amino acids (eg, citrulline), plasma carnitine and acylcarnitine esters, and urine organic acids and orotic acid. Respiratory alkalosis is usually present in urea cycle defects and transient hyperammonemia of the newborn, while acidosis is characteristic of hyperammonemia due to organic acidemias.
6. Acidosis
Inborn errors may cause chronic or acute acidosis at any age, with or without an increased anion gap. Inborn errors should be considered when acidosis occurs with recurrent vomiting or hyperammonemia and when acidosis is out of proportion to the clinical status. Acidosis due to an inborn error can be difficult to correct. The main causes of anion gap metabolic acidosis are lactic acidosis, ketoacidosis (including abnormal ketone body production such as in β-ketothiolase deficiency), methylmalonic aciduria or other organic acidurias, intoxication (ethanol, methanol, ethylene glycol, and salicylate), and uremia. Causes of non–anion gap metabolic acidosis include loss of base in diarrhea or renal tubular acidosis (isolated renal tubular acidosis or renal Fanconi syndrome). If renal bicarbonate loss is found, a distinction must be made between isolated renal tubular acidosis and a more generalized renal tubular disorder or renal Fanconi syndrome by testing for renal losses of phosphorus and amino acids. Inborn errors associated with renal tubular acidosis or renal Fanconi syndrome include cystinosis, tyrosinemia type I, carnitine palmitoyltransferase I, galactosemia, hereditary fructose intolerance, Lowe syndrome, and mitochondrial diseases. Serum glucose and ammonia levels and urinary pH and ketones should be examined. Samples for amino acids and organic acids should be obtained at once and may be evaluated immediately or frozen for later analysis, depending on how strongly an inborn error is suspected. It is useful to test blood lactate and pyruvate levels in the chronically acidotic patient even if urine organic acid levels are normal. Lactate and pyruvate levels are difficult to interpret in the acutely ill patient, but in the absence of shock, high levels of lactic acid suggest primary lactic acidosis.
MANAGEMENT OF METABOLIC EMERGENCIES
Patients with severe acidosis, hypoglycemia, and hyperammonemia may be very ill; initially mild symptoms may worsen quickly, and coma and death may ensue within hours. With prompt and vigorous treatment, however, patients can recover completely, even from deep coma. All oral intake should be stopped. Sufficient glucose should be given intravenously to avoid or minimize catabolism in a patient with a known inborn error who is at risk for crisis. Most conditions respond favorably to glucose administration, although a few (eg, primary lactic acidosis due to pyruvate dehydrogenase deficiency) do not. After exclusion of fatty acid oxidation disorders, immediate institution of intravenous fat emulsions (eg, intralipid) can provide crucial caloric input. Severe or increasing hyperammonemia should be treated pharmacologically or with dialysis (see Disorders of the Urea Cycle), and severe acidosis should be treated with bicarbonate. More specific measures can be instituted when a diagnosis is established.
NEWBORN SCREENING
Criteria for screening newborns for a disorder include its frequency, its consequences if untreated, the ability of therapy to mitigate consequences, the cost of testing, and the cost of treatment. With the availability of tandem mass spectrometry, newborn screening has expanded greatly to now include 20 core conditions and multiple secondary conditions screened by most states. In general, amino acidopathies, organic acidurias, and disorders of fatty acid oxidation are the disorders for which screening now occurs. Most states also screen for hypothyroidism, congenital adrenal hyperplasia, hemoglobinopathies, biotinidase deficiency, galactosemia, and cystic fibrosis. Screening for severe combined immune deficiency has been recently added. Screening should occur for all infants between 24 and 72 hours of life or before hospital discharge.
Some screening tests measure a metabolite (eg, phenylalanine) that becomes abnormal with time and exposure to diet. In such instances, the disease cannot be detected reliably until intake of the substrate is established. Other tests measure enzyme activity and can be performed at any time (eg, for biotinidase deficiency). Transfusions may cause false-negative results in this instance, and exposure of the sample to heat may cause false-positive results. Technologic advances have extended the power of newborn screening but have brought additional challenges. For example, although tandem mass spectrometry can detect many more disorders in the newborn period, consensus on diagnosis and treatment for some conditions is still under development.
Screening tests are not diagnostic, and diagnostic tests must be undertaken when an abnormal screening result is obtained. Further, because false-negative results occur, a normal newborn screening test does not rule out a condition.
The appropriate response to an abnormal screening test depends on the condition in question and the predictive value of the test. For example, when screening for galactosemia by enzyme assay, complete absence of enzyme activity is highly predictive of classic galactosemia. Failure to treat may rapidly lead to death. In this case, treatment must be initiated immediately while diagnostic studies are pending. In phenylketonuria, however, a diet restricted in phenylalanine is harmful to the infant whose screening test is a false-positive, while diet therapy produces an excellent outcome in the truly affected infant if treatment is established within the first weeks of life. Therefore, treatment for phenylketonuria should only be instituted when the diagnosis is confirmed. Physicians should review American College of Medical Genetics recommendations, state laws and regulations, and consult with their local metabolic center to arrive at appropriate strategies for each hospital and practice.
Cook P, Walker V: Investigation of the child with an acute metabolic disorder. J Clin Pathol 2011;64(3):181 [PMID: 21258090].
Ficicioglu C, Bearden D: Isolated neonatal seizures: when to suspect inborn errors of metabolism. Pediatr Neurol 2011;45(5):283 [PMID: 22000307].
Lindner M et al: Efficacy and outcome of expanded newborn screening for metabolic diseases: report of 10 years from South-West Germany. Orphanet J Rare Dis 2011;6:44 [PMID: 21689452].
Michelson DJ, Shevell MI, Sherr EH, Moeschler JB, Gropman AL, Ashwal S: Evidence report: genetic and metabolic testing on children with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2011;77(17):1629 [PMID: 21956720].
Shanmugam NP, Bansal S, Greenough A, Verma A, Dhawan A: Neonatal liver failure: aetiologies and management—state of the art. Eur J Pediatr 2011;170(5):573 [PMID: 20886352].
Sun A, Lam C, Wong DA: Expanded newborn screening for inborn errors of metabolism: overview and outcomes. Adv Pediatr 2012;59(1):209 [PMID: 22789580].
van Karnebeek CD, Stockler S: Treatable inborn errors of metabolism causing intellectual disability: a systematic literature review. Mol Genet Metab 2012;105(3):368 [PMID: 22212131].
Wilcken B: Newborn screening: how are we traveling, and where should we be going? J Inherit Metab Dis 2011;34(3):569 [PMID: 21499716].
DISORDERS OF CARBOHYDRATE METABOLISM
GLYCOGEN STORAGE DISEASES
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Types 0, I, III, VI, and IX manifest with hypoglycemia in infants.
Types 0, I, III, VI, and IX manifest with hypoglycemia in infants.
 Types II, V, and VII manifest with rhabdomyolysis or muscle weakness.
Types II, V, and VII manifest with rhabdomyolysis or muscle weakness.
 Types IV and IX manifest with hepatic cirrhosis.
Types IV and IX manifest with hepatic cirrhosis.
Glycogen is a highly branched polymer of glucose that is stored in liver and muscle. Different enzyme defects affect its biosynthesis and degradation. The hepatic forms of the glycogenoses cause growth failure, hepatomegaly, and severe fasting hypoglycemia. They include glucose-6-phosphatase deficiency (type I; von Gierke disease), debrancher enzyme deficiency (type III), hepatic phosphorylase deficiency (type VI), and phosphorylase kinase deficiency (type IX), which normally regulates hepatic phosphorylase activity. Glycogen synthase deficiency (GSD0) causes hypoglycemia usually after about 12 hours fasting, and can cause mild postprandial hyperglycemia and hyperlactatemia. There are two forms of glucose-6-phosphatase deficiency: in type Ia, the catalytic glucose-6-phosphatase is deficient, and in type Ib, the glucose-6-phosphate transporter is deficient. The latter form also has neutropenia. Glycogenosis type IV, brancher enzyme deficiency, usually presents with progressive liver cirrhosis, as do some rare forms of phosphorylase kinase deficiency.
The myopathic forms of glycogenosis affect skeletal muscle. Skeletal myopathy with weakness or rhabdomyolysis may be seen in muscle phosphorylase deficiency (type V), phosphofructokinase deficiency (type VII), and acid maltase deficiency (type II; Pompe disease). The infantile form of Pompe disease also has hypertrophic cardiomyopathy and macroglossia. The gluconeogenetic disorder fructose-1,6-bisphosphatase deficiency presents with major lactic acidosis and delayed hypoglycemia on fasting.
 Diagnosis
Diagnosis
Initial tests include glucose, lactate, triglycerides, cholesterol, uric acid, transaminases, and creatine kinase. Functional testing includes responsiveness of blood glucose and lactate to fasting; for myopathic forms, an ischemic exercise test is helpful. Most glycogenoses can now be diagnosed by molecular analysis, including next-generation panels. Other diagnostic studies include enzyme assays of leukocytes, fibroblasts, liver, or muscle. Disorders diagnosable from analysis of red or white blood cells include deficiency of debrancher enzyme (type III) and phosphorylase kinase (type IX). Pompe disease can usually be diagnosed by assaying acid maltase in a blood spot with confirmation in fibroblasts.
 Treatment
Treatment
Treatment is designed to prevent hypoglycemia and avoid secondary metabolite accumulations such as elevated lactate in glycogenosis type I. In the most severe hepatic forms, the special diet must be strictly monitored with restriction of free sugars and measured amounts of uncooked cornstarch, which slowly releases glucose in the intestinal lumen. Good results have been reported following continuous nighttime carbohydrate feeding or uncooked cornstarch therapy. Late complications even after years of treatment include focal segmental glomerulosclerosis, hepatic adenoma or carcinoma, and gout. Enzyme-replacement therapy in Pompe disease corrects the cardiomyopathy, but the response in skeletal myopathy is variable with optimal results seen in patients treated early and who have mutations that allow formation of some residual protein which is detected as cross-reacting material on Western blotting. Immunomodulation is used for patients whose treatment response declines due to antibodies to the recombinant enzyme.
Bhattacharya K: Dietary dilemmas in the management of glycogen storage disease type 1. J Inherit Metab Dis 2011;34:621 [PMID: 21491105].
DiMauro S, Spiegel R: Progress and problems in muscle glycogenoses. Acta Myol 2011;30(2):96 [PMID: 22106711].
Hicks J, Wartchow E, Mierau G: Glycogen storage diseases: a brief review and update on clinical features, genetic abnormalities, pathologic features, and treatment. Ultrastruct Pathol 2011;35(5):183 [PMID: 21910565].
Kishnani PS, Beckemeyer AA, Mendelsohn NJ: The new era of Pompe disease: advances in the detection, understanding of the phenotypic spectrum, pathophysiology, and management. Am J Med Genet C Semin Med Genet 2012;160(1):1 [PMID: 22253049].
Kishnani PS et al: Glycogen storage disease type III diagnosis and management guidelines. Genet Med 2010;12:446 [PMID: 20631546].
Patient and parent support group website with useful information for families: http://www.agsdus.org.
Rake JP et al: Guidelines for management of glycogen storage disease type I—European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr 2002;161(Suppl 1):S112 [PMID: 12373584].
Visser G et al: Consensus guidelines for management of glycogen storage disease type 1b—European Study on Glycogen Storage Disease Type 1. Eur J Pediatr 2002;161(Suppl 1):S120 [PMID: 12373585].
GALACTOSEMIA
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Severely deficient neonates present with vomiting, jaundice, and hepatomegaly on initiation of lactose-containing feedings.
Severely deficient neonates present with vomiting, jaundice, and hepatomegaly on initiation of lactose-containing feedings.
 Renal Fanconi syndrome, cataracts of the ocular lens, hepatic cirrhosis, and sepsis occur in untreated children.
Renal Fanconi syndrome, cataracts of the ocular lens, hepatic cirrhosis, and sepsis occur in untreated children.
 Delayed, apraxic speech and ovarian failure occur frequently even with treatment. Developmental delay, tremor, and ataxia occur less frequently.
Delayed, apraxic speech and ovarian failure occur frequently even with treatment. Developmental delay, tremor, and ataxia occur less frequently.
Classic galactosemia is caused by almost total deficiency of galactose-1-phosphate uridyltransferase. Accumulation of galactose-1-phosphate causes hepatic parenchymal disease and renal Fanconi syndrome. Onset of the severe disease is marked in the neonate by vomiting, jaundice (both direct and indirect), hepatomegaly, and rapid onset of liver insufficiency after initiation of milk feeding. Hepatic cirrhosis is progressive. Without treatment, death frequently occurs within a month, often from Escherichia coli sepsis. Cataracts usually develop within 2 months in untreated cases but usually reverse with treatment. With prompt institution of a galactose-free diet, the prognosis for survival without liver disease is excellent. Even when dietary restriction is instituted early, patients with galactosemia are at increased risk for speech and language deficits and ovarian failure. Some patients develop progressive mental retardation, tremor, and ataxia. Milder variants of galactosemia with better prognosis exist.
The disorder is autosomal recessive with an incidence of approximately 1:40,000 live births.
 Diagnosis
Diagnosis
In infants receiving foods containing galactose, laboratory findings include liver dysfunction, particularly PT prolongation, together with proteinuria and aminoaciduria. Absence of reducing substances in urine does not exclude the diagnosis. Galactose-1-phosphate is elevated in red blood cells. When the diagnosis is suspected, galactose-1-phosphate uridyltransferase should be assayed in erythrocytes. Blood transfusions give false-negative results and sample deterioration false-positive results.
Newborn screening by demonstrating enzyme deficiency in red cells with the Beutler test or by demonstrating increased serum galactose allows timely institution of treatment.
 Treatment
Treatment
A galactose-free diet should be instituted as soon as the diagnosis is made. Compliance with the diet must be monitored by following galactose-1-phosphate levels in red blood cells. Appropriate diet management requires not only the exclusion of milk but an understanding of the galactose content of foods. Avoidance of galactose should be lifelong with appropriate calcium replacement, intake of which tends to be low due to the restriction of dairy products.
Berry GT: Galactosemia: when is it a newborn screening emergency? Mol Genet Metab 2012;106(1):7 [PMID: 22483615].
Berry GT, Elsas LJ: Introduction to the Maastricht workshop: lessons from the past and new directions in galactosemia. J Inherit Metab Dis 2011;34(2):249 [PMID: 21116719].
Bosch AM: Classic galactosemia: dietary dilemmas. J Inherit Metab Dis 2011;34(2):257 [PMID: 20625932].
Fridovich-Keil JL, Gubbels CS, Spencer JB, Sanders RD, Land JA, Rubio-Gozalbo E: Ovarian function in girls and women with GALT-deficiency galactosemia. J Inherit Metab Dis 2011;34(2):357 [PMID: 20978943].
Hoffmann B, Wendel U, Schweitzer-Krantz S: Cross-sectional analysis of speech and cognitive performance in 32 patients with classic galactosemia. J Inherit Metab Dis 2011;34(2):421 [PMID: 21347587].
Patient and parent support group website with useful information for families: http://www.galactosemia.org.
Timmers I, van den Hurk J, Di Salle F, Rubio-Gozalbo ME, Jansma BM: Language production and working memory in classic galactosemia from a cognitive neuroscience perspective: future research directions. J Inherit Metab Dis 2011;34(2):367 [PMID: 21290187].
HEREDITARY FRUCTOSE INTOLERANCE
Hereditary fructose intolerance is an autosomal recessive disorder in which deficient activity of fructose-1-phosphate aldolase causes hypoglycemia and tissue accumulation of fructose-1-phosphate on fructose ingestion. Other abnormalities include failure to thrive, vomiting, jaundice, hepatomegaly, proteinuria, and renal Fanconi syndrome. The untreated condition can progress to death from liver failure.
 Diagnosis
Diagnosis
The diagnosis is suggested by finding fructosuria or an abnormal transferrin glycoform in the untreated patient. Diagnosis is made by sequencing the HFI gene. An alternative method for diagnosis is enzyme assay of fructose-1-phosphate aldolase in liver biopsy.
 Treatment
Treatment
Treatment consists of strict dietary avoidance of fructose. Vitamin supplementation is usually needed. Drugs and vitamins dispensed in a sucrose base should be avoided. Treatment monitoring can be done with transferrin glycoform analysis. If diet compliance is poor, physical growth retardation may occur. Growth will resume when more stringent dietary restrictions are reinstituted. If the disorder is recognized early, the prospects for normal development are good. As affected individuals grow up, they may recognize the association of nausea and vomiting with ingestion of fructose-containing foods and selectively avoid them.
Bouteldja N, Timson DJ: The biochemical basis of hereditary fructose intolerance. J Inherit Metab Dis 2010;33(2):105 [PMID: 20162364].
Website with useful information for families: http://www.bu.edu/aldolase.
DISORDERS OF ENERGY METABOLISM
The most common disorders of central mitochondrial energy metabolism are pyruvate dehydrogenase deficiency and deficiencies of respiratory chain components. Disorders of the Krebs cycle include deficiencies in fumarase, 2-ketoglutarate dehydrogenase, and succinyl-CoA ligase. In many, but not in all, patients lactate is elevated in either blood or CSF. In pyruvate dehydrogenase deficiency, the lactate-pyruvate ratio is normal, whereas in respiratory chain disorders the ratio is often increased. Care must be taken to distinguish an elevated lactate level that is due to these conditions (called primary lactic acidoses) from elevated lactate that is a consequence of hypoxia, ischemia, or sampling problems. Table 36–3 lists some causes of primary lactic acidosis.
Table 36–3. Causes of primary lactic acidosis in childhood.
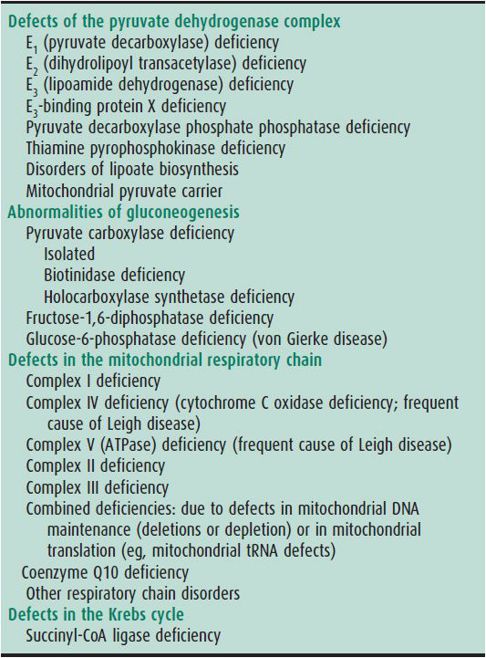
Patients with a defect in the pyruvate dehydrogenase complex often have agenesis of the corpus callosum or Leigh syndrome (lesions in the globus pallidus, dentate nucleus, and periaqueductal gray matter). They can have mild facial dysmorphism. Recurrent altered mental status, recurrent ataxia, and recurrent acidosis are typical of many disturbances of pyruvate metabolism. The most common genetic defect is in the X-linked E1-α component, with males carrying milder mutations and females carrying severe mutations leading to periventricular cystic brain lesions.
The respiratory chain disorders are frequent (1:5000) and involve a heterogenous group of genetic defects that produce a variety of clinical syndromes (now > 50) of varying severity and presentation. The disorders can affect multiple organs. The following set of symptoms (not intended as a comprehensive listing) can indicate a respiratory chain disorder:
2. Brain: Progressive neurodegeneration, Leigh syndrome, myoclonic seizures, brain atrophy, and subcortical leukodystrophy
3. Eye: Optic neuropathy, retinitis pigmentosa, progressive external ophthalmoplegia, and cataracts
4. Ears: Nerve deafness
5. Muscle: Myopathy with decreased endurance or rhabdomyolysis
6. Kidney: Renal Fanconi syndrome, proteinuria (in CoQ deficiency)
7. Endocrine: Diabetes mellitus and hypoparathyroidism
8. Intestinal: Pancreatic or liver insufficiency or pseudoobstruction
9. Skin: Areas of hypopigmentation
10. Heart: Cardiomyopathy, conduction defects, and arrhythmias
Respiratory chain disorders are among the more common causes of static, progressive, or self-limited neurodevelopmental problems in children. Patients may present with nonspecific findings such as hypotonia, failure to thrive, or renal tubular acidosis, or with more specific features such as ophthalmoplegia or cardiomyopathy. Symptoms are often combined in recognizable clinical syndromes with ties to specific genetic causes. Ragged red fibers and mitochondrial abnormalities may be noted on histologic examination of muscle. Thirteen of the more than 100 genes that control activity of the respiratory chain are part of the mitochondrial genome. Therefore, inheritance of defects in the respiratory chain may be mendelian or maternal.
 Diagnosis
Diagnosis
Pyruvate dehydrogenase deficiency is diagnosed by enzyme assay in leukocytes or fibroblasts. Confirmation can be obtained by molecular analysis. Diagnosis of respiratory chain disorders is based on a convergence of clinical, biochemical, morphologic, enzymatic, and molecular data. Classic pathologic features of mitochondrial disorders are the accumulation of mitochondria, which produces ragged red fibers in skeletal muscle biopsy, and abnormal mitochondrial shapes and inclusions inside mitochondria on electron microscopy. However, these findings are only present in 5% of children. Enzyme analysis on skeletal muscle or liver tissue is complicated by an overlap between normal and affected range. Blue native polyacrylamide gel electrophoresis (PAGE) analyzes the assembly of the respiratory chain enzyme complexes. Mitochondrial DNA (mtDNA) analysis in blood or tissue may identify a diagnostic mutation. A rapidly increasing number of nuclear genes (now > 100) causing respiratory chain defects are recognized. Children with defects in mtDNA maintenance (such as mtDNA polymerase γ, POLG1 gene) often present with liver disease and neurodegeneration and are diagnosed by sequencing the causative nuclear genes. Next-generation sequencing of panels of genes or exome sequencing is increasingly used due to the large number of genes involved, but functional testing is often required for confirmation of the pathogenicity of identified mutations. Although diagnostic criteria have been published, the cause of respiratory chain defects still cannot be defined in many patients. In some instances, the genetics and prognosis may be clear, but in many cases neither prognosis nor genetic risk can be predicted. Because of the high complexity of this group of disorders, many patients require a high degree of expertise and multiple studies to arrive at a final diagnosis.
 Treatment
Treatment
A ketogenic diet is useful in pyruvate dehydrogenase deficiency. In rare patients with primary coenzyme Q deficiency, coenzyme Q treatment is very effective. Other treatments are of theoretical value, with little data on efficacy. Thiamine and lipoic acid have been tried in patients with pyruvate dehydrogenase complex deficiencies, and coenzyme Q and riboflavin have been helpful in some patients with respiratory chain defects. Dichloroacetic acid has limited clinical response in pyruvate dehydrogenase deficiency and has adverse effects. Avoidance of catabolism and of medications that impair mitochondrial function is an important component of treating patients with respiratory chain defects. Transcriptional upregulation in partial deficiencies of nuclear origin and new antioxidants such as EPI-743 offer new hope for the treatment of respiratory chain defects.
Davis RL, Sue CM: The genetics of mitochondrial disease. Semin Neurol 2011;31(5):519 [PMID: 22266889].
Koopman WJH, Willems PHGM, Smeitink JAM: Monogenic mitochondrial disorders. New Engl J Med 2012;366:1132–1141 [PMID: 22435372]
Kisler JE et al: Mitochondrial disease in childhood: a clinical approach to investigation and management. Dev Med Child Neurol 2010;52:422 [PMID: 20163433].
Marin-Valencia I et al: Pyruvate carboxylase deficiency: mechanisms, mimics and anaplerosis. Mol Genet Metab 2010;101:9 [PMID: 20598931].
Patel KP, O’Brien TW, Subramony SH, Shuster J, Stacpoole PW: The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab 2012;105(1):34 [PMID: 22079328].
Patient and parent support group website with useful information for families: http://www.umdf.org.
Scaglia F: Nuclear gene defects in mitochondrial disorders. Methods Mol Biol 2012;837:17 [PMID: 22215538].
Spinazzola A: Mitochondrial DNA mutations and depletion in pediatric medicine. Semin Fetal Neonatal Med 2011;16(4):190 [PMID: 21652274].
Wong LJ: Mitochondrial syndromes with leukoencephalopathies. Semin Neurol 2012;32(1):55 [PMID: 22422207].
DISORDERS OF AMINO ACID METABOLISM
DISORDERS OF THE UREA CYCLE
Ammonia is derived from the catabolism of amino acids and is converted to an amino group in urea by enzymes of the urea cycle. Patients with severe defects (often those enzymes early in the urea cycle) usually present in infancy with severe hyperammonemia, vomiting, and encephalopathy, which is rapidly fatal. Patients with milder genetic defects may present with vomiting, encephalopathy, or liver failure after increased protein ingestion or infection. Although defects in argininosuccinic acid synthetase (citrullinemia) and argininosuccinic acid lyase (argininosuccinic acidemia) may cause severe hyperammonemia in infancy, the usual clinical course is chronic with mental retardation. Ornithine transcarbamylase deficiency is X-linked; the others are autosomal recessive. Age at onset of symptoms varies with residual enzyme activity, protein intake, growth, and stresses such as infection. Even within a family, males with ornithine transcarbamylase deficiency may differ by decades in the age of symptom onset. Many female carriers of ornithine transcarbamylase deficiency have protein intolerance. Some develop migraine-like symptoms after protein loads, and others develop potentially fatal episodes of vomiting and encephalopathy after protein ingestion, infections, or during labor and delivery. Trichorrhexis nodosa is common in patients with argininosuccinic aciduria.
 Diagnosis
Diagnosis
Blood ammonia should be measured in any acutely ill newborn in whom a cause is not obvious. In urea cycle defects, early hyperammonemia is associated with hyperventilation and respiratory alkalosis. Serum citrulline is low or undetectable in carbamoyl phosphate synthetase and ornithine transcarbamylase deficiency, high in argininosuccinic acidemia, and very high in citrullinemia. Large amounts of argininosuccinic acid are found in the urine of patients with argininosuccinic acidemia. Urine orotic acid is increased in infants with ornithine transcarbamylase deficiency. Citrullinemia and argininosuccinic acidemia can be diagnosed in utero by appropriate enzyme assays in uncultured chorionic villus sample (CVS) material or in amniocytes, but carbamoyl phosphate synthetase and ornithine transcarbamylase deficiency can be diagnosed in utero only by molecular methods.
 Treatment
Treatment
During treatment of acute hyperammonemic crisis, protein intake should be stopped, and glucose and lipids should be given to reduce endogenous protein breakdown from catabolism. Careful administration of essential amino acids facilitates protein anabolism. Arginine is given intravenously. It is an essential amino acid for patients with urea cycle defects and increases the excretion of waste nitrogen in citrullinemia and argininosuccinic acidemia. Sodium benzoate and sodium phenylacetate are given intravenously to increase excretion of nitrogen as hippuric acid and phenylacetylglutamine. Additionally, hemodialysis or hemofiltration is indicated for severe or persistent hyperammonemia, as is usually the case in the newborn. Peritoneal dialysis and exchange transfusion are ineffective. Long-term treatment includes low-protein diet, oral administration of arginine or citrulline, and sodium benzoate or sodium phenylbutyrate (a prodrug of sodium phenylacetate). Symptomatic heterozygous female carriers of ornithine transcarbamylase deficiency should also receive such treatment. Liver transplantation may be curative and is indicated for patients with severe disorders.
The outcome of urea cycle disorders depends on the genetic severity of the condition (residual activity) and the severity and prompt treatment of hyperammonemic episodes. Brain damage depends on the duration and the degree of elevation of ammonia (and glutamine). Many patients with urea cycle defects, no matter what the enzyme defect, develop permanent neurologic and intellectual impairments, with cortical atrophy and ventricular dilation seen on computed tomographic scan. Rapid identification and treatment of the initial hyperammonemic episode improve outcome.
Auron A, Brophy PD: Hyperammonemia in review: pathophysiology, diagnosis, and treatment. Pediatr Nephrol 2012;27(2):207 [PMID: 21431427].
Bireley WR, Van Hove JL, Gallagher RC, Fenton LZ: Urea cycle disorders: brain MRI and neurological outcome. Pediatr Radiol 2012:42(4):455 [PMID: 21989980].
Braissant O: Current concepts in the pathogenesis of urea cycle disorders. Mol Genet Metab 2010;100(Suppl 1):S3 [PMID: 20227314].
Darwish AA, McKiernan P, Chardot C: Paediatric liver transplantation for metabolic disorders. Part 1: Liver-based metabolic disorders without liver lesions. Clin Res Hepatol Gastroenterol 2011;35(3):194 [PMID: 21376697].
Häberle J: Clinical practice: the management of hyperammonemia. Eur J Pediatr 2011;170(1):21 [PMID: 21165747].
Patient and parent support group website with useful information for families: http://www.nucdf.org.
Scaglia F: New insights in nutritional management and amino acid supplementation in urea cycle disorders. Mol Genet Metab 2010;100(Suppl 1):S72 [PMID: 20299258].
Summar ML et al: Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year, multicentre study of acute hyperammonaemic episodes. Acta Paediatr 2008;97:1420 [PMID: 18647279].
Urea Cycle Disorders Consortium: http://rarediseasesnetwork.epi.usf.edu/ucdc/about/index.htm.
PHENYLKETONURIA & THE HYPERPHENYLALANINEMIAS
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Mental retardation, hyperactivity, seizures, light complexion, and eczema characterize untreated patients.
Mental retardation, hyperactivity, seizures, light complexion, and eczema characterize untreated patients.
 Newborn screening for elevated serum phenylalanine identifies most infants.
Newborn screening for elevated serum phenylalanine identifies most infants.
 Disorders of cofactor metabolism also produce elevated serum phenylalanine level.
Disorders of cofactor metabolism also produce elevated serum phenylalanine level.
 Early diagnosis and treatment with phenylalanine-restricted diet prevents mental retardation.
Early diagnosis and treatment with phenylalanine-restricted diet prevents mental retardation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


