 Figure 33–1. General approach to primary immunodeficiencies.
Figure 33–1. General approach to primary immunodeficiencies.
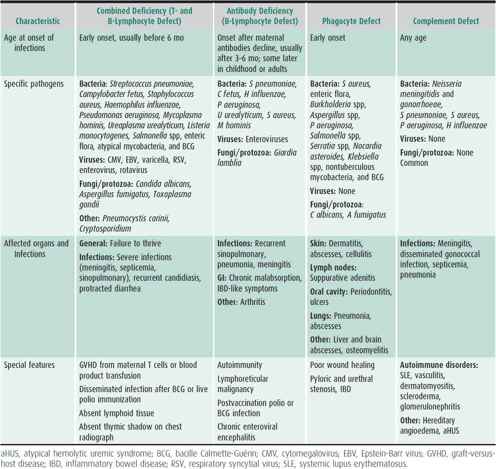
Key clinical patterns can indicate the presence of a PID and the category of immune impairment. Frequency, severity, and age of onset of infections are important clues. The Modell Foundation warning signs for PID are shown in Figure 33–2. Children who meet two or more of these signs should be screened for PID. The type of infections should guide initial investigation, as antibody, complement, and phagocyte defects predispose mainly to bacterial infections, but diarrhea, superficial candidiasis, opportunistic infections, and severe herpesvirus infections are more characteristic of T-lymphocyte immunodeficiency. Combined immunodeficiency syndromes will present with a combination of infections typical for B- and T-lymphocyte deficiencies. Table 33–1 classifies PID into four main host immunity categories based on age of onset, infections with specific pathogens, affected organs, and other special features. Finally, male gender increases the likelihood of an X-linked (XL) PID, while consanguinity increases the likelihood of an autosomal recessive (AR) form of PID.
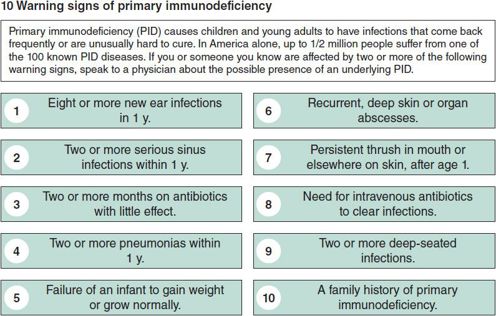
 Figure 33–2. Warning signs of primary immunodeficiency. (Data from the Jeffrey Modell Foundation.)
Figure 33–2. Warning signs of primary immunodeficiency. (Data from the Jeffrey Modell Foundation.)
Table 33–1. Clinical features of primary immunodeficiencies.
Research on deficiencies of the innate immune response is an evolving field. Pattern-recognition receptors (PRRs) are important for the recognition of pathogen-associated molecular patterns specific for different microbes, initiation of the innate immune response, and cross talk with adaptive immunity. They are expressed on the surface or in the cytoplasm of cells of the innate immune system, dependent on where specific microbes are encountered. Four classes of PRRs have been identified. Toll-like receptors (TLRs) recognize a spectrum of bacteria, viruses, selected fungi, and protozoa. C-type lectin receptors (CLRs) that include dectin-1 and mannose-binding lectin are involved in recognition of bacteria and fungi. Cytoplasmic PRRs include nucleotide-binding oligomerization domain (NOD) leucine-rich-repeat containing receptors (NLRs) that recognize peptidoglycan structures on bacteria, and retinoic acid-inducible gene I protein (RIG-1) helicase receptors that recognize viral nucleic acids. Dependent on the defect in PRR signaling, patients may present with an increased susceptibility of bacterial, viral, or fungal infections.
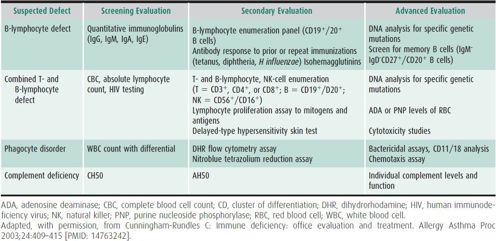
Laboratory investigation should be directed by the clinical presentation and the suspected category of host immunity impairment. A complete blood cell count (CBC) with cell differential and measurement of quantitative immunoglobulins (Igs) will identify the majority of patients with PID, as antibody deficiencies account for at least 50% of PIDs (Figure 33–3). Table 33–2 summarizes the approach to laboratory evaluation of PID.
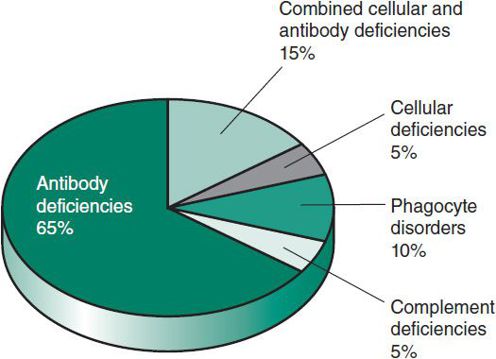
 Figure 33–3. Relative frequencies of primary immunodeficiencies. (Adapted, with permission, from Stiehm ER et al (eds): Immunologic Disorders in Infants and Children, 5th ed. Elsevier; 2004.)
Figure 33–3. Relative frequencies of primary immunodeficiencies. (Adapted, with permission, from Stiehm ER et al (eds): Immunologic Disorders in Infants and Children, 5th ed. Elsevier; 2004.)
Table 33–2. Laboratory evaluation for primary immunodeficiency.
Antibodies & Immunoglobulins
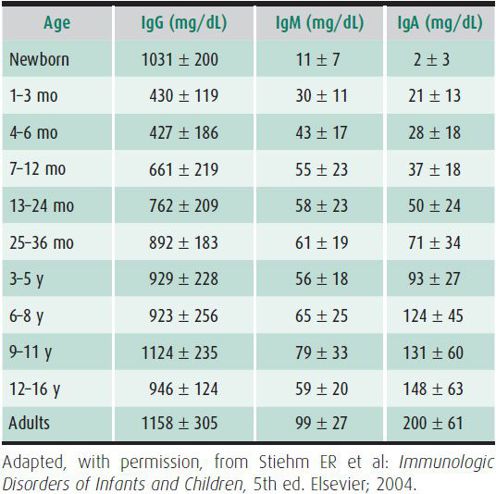
The initial laboratory screening for antibody deficiency includes the measurement of serum Igs: IgG, IgM, IgA, and IgE, which have age-dependent normal ranges (Table 33–3). Normal IgG, IgM, and IgA and increased IgE levels are indicative of atopy. Some patients may have normal Ig levels but fail to make protective antibodies to certain microbes; other patients have subnormal Ig levels but make protective antibodies. Specific antibodies include isohemagglutinins, naturally occurring IgM antibodies that are detectable by age 6 months except in children with blood group AB. Specific IgG antibodies to protein antigens (tetanus, diphtheria, rubella, mumps) and polysaccharide antigens (Haemophilus influenzae, Streptococcus pneumoniae) can be measured after immunization. The response to polysaccharide antigens develops during the second year of life, but protein-conjugated vaccines elicit an earlier response in immunocompetent children. Assessing the antibody response to pneumococcal polysaccharide antigens can be helpful in the face of repeated pneumococcal infections. The gold standard is comparison of pre- and postimmunization titers.
Table 33–3. Normal values for immunoglobulins by age.
Obtaining a CBC with differential and T- and B-lymphocyte counts are recommended if an initial screen reveals very low concentrations of all Ig classes. Certain types of hypogammaglobulinemia are characterized by low levels of or absent B lymphocytes, such as XL Bruton agammaglobulinemia. Protein electrophoresis can help identify monoclonal gammopathy as seen in XL lymphoproliferative syndrome, which can be complicated by fatal Epstein-Barr virus (EBV) infection, and in heavy-chain diseases. Serum albumin should be measured in patients with hypogammaglobulinemia to exclude secondary deficiencies due to protein loss through bowel or kidneys. IgG or IgA subclass measurements may be abnormal in patients with varied immunodeficiency syndromes, but they are rarely helpful in an initial evaluation.
T Lymphocytes
The initial laboratory screening for a T-lymphocyte deficiency includes a CBC with cell differential to evaluate for a decreased absolute lymphocyte count (< 1000/μL) and enumeration of absolute numbers of T cells and their subsets, B cells, and natural killer (NK) cells (see Table 33–2). T-cell function can be analyzed by in vitro lymphocyte proliferation assays to mitogens that stimulate all T cells and by specific antigens that stimulate only antigen-specific T cells. Borderline function must be interpreted based on clinical correlation. T-lymphocyte function is often also studied in vivo by delayed hypersensitivity skin tests to specific antigens, including Candida albicans, tetanus, or mumps, but a negative result is not helpful, as it may be due to young age, chronic illness, vitamin D deficiency, or poor test technique. T-lymphocyte deficiencies will often not manifest as skin-test anergy until the impairment is severe, for example as in AIDS. It is important to evaluate a patient’s specific antibody production because proper B-lymphocyte function and antibody production are dependent on adequate T-lymphocyte function. Therefore, most T-lymphocyte deficiencies manifest as combined T- and B-lymphocyte deficiencies.
Phagocyte Immunity
The initial laboratory screening for phagocyte disorders, mainly impaired neutrophil function, should include a CBC and cell differential to look for neutropenia. A blood smear can detect Howell-Jolly bodies in erythrocytes, indicative of asplenia, and abnormalities in lysosomal granules in neutrophils. An abnormality of the neutrophil respiratory burst, which would lead to impaired neutrophil bactericidal activity, can be tested by nitroblue tetrazolium (NBT) reduction. The dihydrorhodamine (DHR) flow cytometry assay assesses the same function more quantitatively. Leukocyte adhesion molecules can be studied by flow cytometry. Assays to study neutrophil phagocytosis of bacteria and phagocytic microbicidal activity are available in specialized laboratories. The clinical symptom pattern that suggests a possible defect of phagocytic cell function should dictate which tests are used.
Complement Pathways (Figure 33–4)
Testing for total hemolytic complement activity with the CH50 assay screens for most of the diseases of the complement system. A normal CH50 titer depends on the ability of all 11 components of the classic pathway and membrane attack complex to interact and then lyse antibody-coated sheep erythrocytes. Alternative complement pathway deficiencies are identified by subnormal lysis of rabbit erythrocytes in the AH50 assay. For both assays the patient’s serum must be separated and frozen at –70°C within 30–60 minutes after collection to prevent loss of activity. Measuring levels of individual components is not necessary when both CH50 and AH50 are normal. If both the CH50 and AH50 are low, a deficiency in their shared terminal pathway (C3, C5, C6, C7, C8, or C9) would be the most common explanation. If the CH50 is low but the AH50 is normal, the deficiency must affect C1, C4, C2, or components of the lectin pathway. If the AH50 is low but the CH50 is normal, a deficiency in factors D or B or properdin should be suspected.
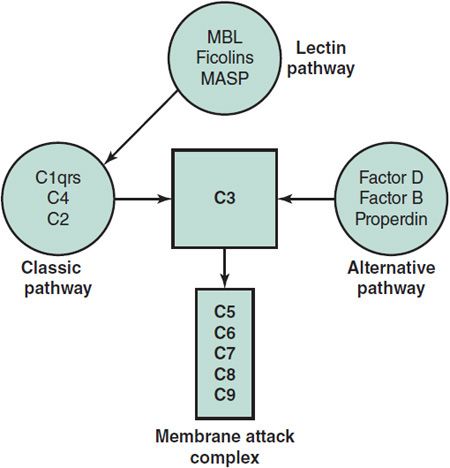
 Figure 33–4. Pathways of complement activation and the central functional role of C3. MASP, MBL-associated serine protease; MBL, mannose-binding lectin.
Figure 33–4. Pathways of complement activation and the central functional role of C3. MASP, MBL-associated serine protease; MBL, mannose-binding lectin.
Pattern-Recognition Receptors
Selected laboratories offer tests to assess TLR function upon stimulation with ligands for different TLRs that have been identified. Mannose-binding lectin levels can be measured in clinical laboratories. However, expression or function of pattern recognition receptors (PRRs) and associated gene mutations are studied mostly in a research setting at the current time.
ANTIBODY DEFICIENCY SYNDROMES
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Recurrent bacterial infections, typically due to encapsulated pyogenic bacteria.
Recurrent bacterial infections, typically due to encapsulated pyogenic bacteria.
 Low immunoglobulin levels.
Low immunoglobulin levels.
 Inability to make specific antibodies to vaccine antigens or infections.
Inability to make specific antibodies to vaccine antigens or infections.
 General Considerations
General Considerations
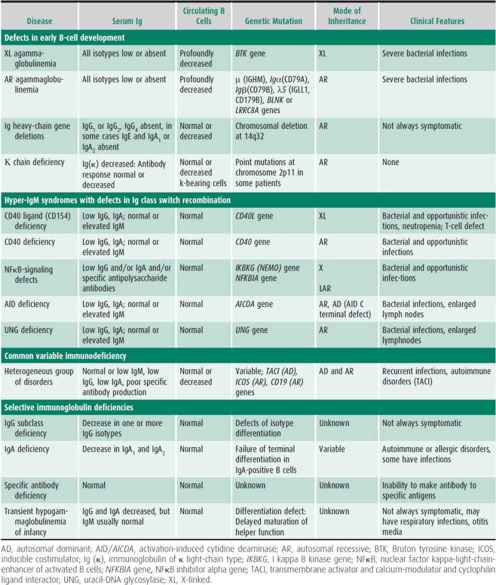
Antibody deficiency syndromes include both congenital and acquired forms of hypogammaglobulinemia with low levels of one or more of the immunoglobulins IgM, IgG, and IgA. Deficiencies result in recurrent bacterial infections, typically with encapsulated bacteria, including pneumonia, otitis, sinusitis, meningitis, cellulitis, and sepsis. As a group, antibody deficiencies represent nearly half of all PIDs. They can be divided into (1) early defects of B-cell development, with absent B cells and severe hypogammaglobulinemia; (2) hyper-IgM syndromes with defects in Ig class switching; (3) common variable immunodeficiency (CVID), with insufficient antibody production; and (4) specific antibody deficiencies. Table 33–4 outlines primary antibody deficiency syndromes, laboratory findings, and genetic inheritance in these disorders.
Table 33–4. Antibody deficiency disorders.
1. X-Linked Agammaglobulinemia
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Infections early in life, usually after age 4 months.
Infections early in life, usually after age 4 months.
 Typical bacterial infections include infections with encapsulated bacteria, for example, H influenzae, S pneumoniae , Staphylococcus aureus, and Pseudo-monas aeruginosa, but also Mycoplasma species.
Typical bacterial infections include infections with encapsulated bacteria, for example, H influenzae, S pneumoniae , Staphylococcus aureus, and Pseudo-monas aeruginosa, but also Mycoplasma species.
 Risk for severe enteroviral infections and, rarely, polio due to live polio vaccine.
Risk for severe enteroviral infections and, rarely, polio due to live polio vaccine.
 Failure to thrive and absent lymphoid tissue on examination.
Failure to thrive and absent lymphoid tissue on examination.
 Very low levels of Igs and B lymphocytes.
Very low levels of Igs and B lymphocytes.
 General Considerations
General Considerations
X-linked agammaglobulinemia (XLA) accounts for 85% of congenital hypogammaglobulinemia and occurs in about 1:200,000 male births. Children with XLA typically present with infections after 4 months of age, when maternally derived IgG levels have declined. XLA is caused by a gene mutation on the X chromosome (Xq21.33–q22) that affects the expression of a B-cell–specific tyrosine kinase (BTK), halting the maturation of B cells and resulting in low or absent B-cell numbers and serum Igs. Early detection and diagnosis of XLA allow Ig-replacement therapy to start before a potentially life-threatening infection can occur.
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Sinopulmonary infections due to H influenzae and S pneumoniae are common, but deep tissue infections and arthritis due to Mycoplasma or Ureaplasma species can occur. Recurrent pulmonary infections may lead to bronchiectasis. Antibody-deficient patients are also at risk for polio due to oral polio vaccine strains, resulting in paralysis, and echoviruses causing chronic encephalitis. At presentation, male infants have scant or absent lymphoid tissue, including tonsils, adenoids, and lymph nodes. A small proportion also has a history of poor growth.
B. Laboratory Findings
Most patients have low levels of or absent Igs M, G, A, and E, and, despite a normal leukocyte count, few or no B lymphocytes. T-lymphocyte numbers and function are normal. Genetic testing for a BTK gene mutation confirms the diagnosis in affected males. Female carriers can be detected by genetic testing or screening for BTK protein expression in platelets followed by mutation analysis if needed.
 Differential Diagnosis
Differential Diagnosis
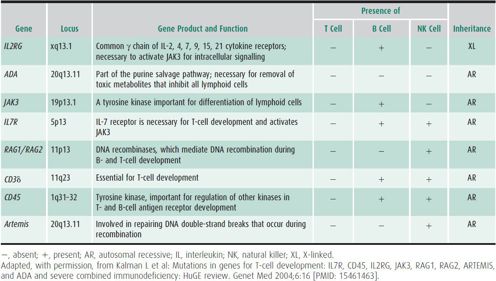
The differential diagnosis includes other causes of antibody deficiency and combined immunodeficiencies (see Table 33–4; Table 33–5). Additional causes of recurrent infections and low Ig levels include protein loss through renal or gastrointestinal disease, but patients with these disorders present with normal numbers of B lymphocytes and, typically, an isolated IgG deficiency.
Table 33–5. Severe combined immunodeficiency variants.
 Treatment
Treatment
Current therapy consists of lifelong Ig-replacement therapy. In addition to preventing infections, Ig replacement usually results in resolution of inflammatory arthritis and improves growth. Because the severity of infections varies and antibiotics are widely used, the diagnosis is often delayed for years, but XLA should be considered in males with recurrent infections regardless of severity. Early diagnosis can prevent permanent disability and premature death.
2. Autosomal Recessive Congenital Agammaglobulinemia
 General Considerations
General Considerations
Autosomal recessive congenital agammaglobulinemia is rare, accounting for less than 15% of all congenital hypogammaglobulinemia, and occurs in both male and female children. In the most common form, it is caused by mutations of the IgM heavy-chain gene on chromosome 14q32. These mutations result in abnormal or absent IgM expression and abnormal development of B cells with decreased or absent antibody production.
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Similarly to patients with XLA, children present with recurrent and severe bacterial infections, typically before age 6 months. Infections include pneumonia, otitis, sinusitis, meningitis, cellulitis, and sepsis. Chronic central nervous system (CNS) infections by enteroviruses have been observed.
B. Laboratory Findings
Patients usually have low numbers of circulating B lymphocytes and low levels of or absent immunoglobulins. Specific antibody function is poor. When the diagnosis is suspected, the detection of a mutation in the μ heavy chain can confirm the most common type. Additional mutations include mutations of the genes encoding the Igα and Igβ molecules, the λ5 surrogate light chain, BLNK or LRRC8 genes.
 Differential Diagnosis
Differential Diagnosis
The differential diagnosis is similar to that of XLA and includes XLA for male patients.
 Treatment
Treatment
Treatment and prognosis are similar to those outlined for XLA.
3. Hyper-IgM Syndromes
Hyper-IgM (HIGM) syndromes are a heterogeneous group of genetic disorders (see Table 33–4) with impaired Ig class switching from IgM to production of IgG, IgA, or IgE associated with normal or elevated serum IgM. If signaling through CD40 is affected, patients present also with opportunistic bacterial infections. Deficiencies of AID or UNG differ from CD40L and CD40 deficiencies in that the patients have large lymph nodes with germinal centers and are not susceptible to opportunistic infections. Patients with HIGM syndrome are at increased risk of autoimmune diseases. Treatment with Ig replacement decreases infections and often normalizes IgM levels. XL CD40L deficiency and NFκB signaling defects will be addressed further in section Other Combined Immunodeficiency Disorders.
4. Common Variable Immunodeficiency
 General Considerations
General Considerations
Common variable immunodeficiency (CVID) is a diagnosis of exclusion after other causes of hypogammaglobulinemia have been eliminated. The onset may be at any age, and the incidence approaches 1:30,000. Many cases are sporadic, but a small percentage of patients have autosomal dominant or recessive inheritance, and some cases are associated with specific HLA-DR/DQ alleles (see Table 33–4).
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Patients have recurrent infections, most often of the sinopulmonary tract, but chronic gastrointestinal infections may manifest with recurrent diarrhea. Patients with CVID are at risk for developing bronchiectasis, autoimmune diseases (idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, rheumatoid arthritis, and inflammatory bowel disease), and malignancies (especially gastric carcinoma and lymphoma).
B. Laboratory Findings
Laboratory findings are variable, but typically reveal low levels of IgG and IgA, normal numbers of B lymphocytes, low numbers of memory B lymphocytes (evaluated by flow cytometry), and abnormal specific antibody levels and responses. Some patients have evidence of T-lymphocyte abnormalities as well. Chronic gastrointestinal tract infections are often due to G lamblia or C jejuni.
Although CVID is typically a diagnosis of exclusion, recent research has revealed multiple specific genetic mutations in patients with CVID. One example is a mutation in a member of the tumor necrosis factor receptor family, identified as transmembrane activator and calcium-modulator and cyclophilin ligand interactor (TACI), which mediates isotype switching in B lymphocytes. TACI mutations were recently found in 10%–15% of patients with CVID, as well as in some relatives of CVID patients with IgA deficiency. The mutations appear to be autosomal dominant, with variable penetrance of clinical immunodeficiency and autoimmune disease, and impairment of T-regulatory cell function.
 Differential Diagnosis
Differential Diagnosis
When a patient presents with recurrent infections, low immunoglobulin levels, and potentially autoimmune symptoms, many different diagnoses must be considered, including other causes of low immunoglobulin levels (loss and abnormal production) and autoimmune diseases. CVID patients have normal numbers of B lymphocytes despite their poor specific antibody responses, which differentiates them from patients with XLA and AR agammaglobulinemia. Patients with CVID also lack the specific mutations responsible for those other disorders. However, CVID patients often have decreased numbers of memory B cells (IgM−/IgD−/CD27+/CD20+ B cells).
 Treatment
Treatment
Treatment includes lifelong Ig-replacement therapy and routine assessment for bronchiectasis, autoimmune disorders, and malignancies. The prognosis can be good and depends on the time to diagnosis and implementation of Ig-replacement therapy. Other complications include B-cell hyperplasia in the gut that may be severe enough to resemble Crohn disease, and gastric atrophy with achlorhydria, sometimes followed by pernicious anemia. Lymphoreticular proliferation can occur after EBV infection and is not always malignant.
5. Acquired Hypogammaglobulinemia
Acquired forms of hypogammaglobulinemia are common and may develop at any age. Causes of secondary hypogammaglobulinemia (nephrotic syndrome and protein-losing enteropathy) should be excluded by measuring serum albumin. The loss of albumin (MW 69323 Da) is usually paralleled by a loss of IgG (MW 150000 Da). Generally, acquired forms are not treated with Ig-replacement therapy because, although immunoglobulin levels are low, antibody function is adequately protective. Morphologic disorders or associated syndromes may point to a specific diagnosis.
6. Transient Hypogammaglobulinemia
Serum IgG levels normally decrease during an infant’s first 4–6 months of life as maternal IgG transmitted in utero is metabolized. Transient hypogammaglobulinemia represents a delay in the onset of immunoglobulin synthesis that results in a prolonged nadir. Symptomatic patients present with recurrent infections, including upper respiratory tract infections, otitis, and sinusitis. The diagnosis is suspected in infants and young children with low levels of IgG and IgA (usually two standard deviations below normal for age), but normal levels of IgM and normal numbers of circulating B lymphocytes. Most affected children have normal specific antibody responses and T-lymphocyte function. Apart from appropriate antibiotics, no treatment is required. Infants with severe infections and hypogammaglobulinemia could be given Ig replacement, but benefits and risk must be considered and this is rarely necessary. Recovery occurs between 18 and 30 months of age, and the prognosis for affected children is excellent provided infections are treated promptly and appropriately.
7. Selective Immunoglobulin Deficiencies
Selective IgA deficiency is the most common immune abnormality, found in approximately 1:700 persons. It is defined by a serum IgA level less than 7 mg/dL. Serum IgM, IgG, specific antibodies, and B- and T-lymphocyte numbers and function are normal. IgA is primarily effective in its secreted form on mucosal surfaces. Therefore, symptomatic patients with low serum IgA develop upper respiratory tract infections, diarrhea, or both, but most people are asymptomatic.
Associations also exist with inflammatory bowel disease, allergic disease, asthma, and autoimmune disorders (thyroiditis, arthritis, vitiligo, thrombocytopenia, and diabetes). IgA replacement is currently not feasible. For the majority of symptomatic IgA-deficient patients, antibiotics and appropriate autoimmune therapies are sufficient. Caution must be exercised, as IgA-deficient patients are at risk for developing anti-IgA antibodies with blood product exposure, and the administration of blood products can result in anaphylaxis. Therefore, when blood products are needed, washed packed red blood cells and volume expanders without blood products are recommended.
The possibility that deficiency of an IgG subclass (eg, abnormally low serum IgG2, IgG3, or IgG4) might predispose to recurrent upper respiratory tract infections in patients with normal total serum immunoglobulin levels is not well established. Normally, IgG1 comprises over 60% of total IgG, IgG2 over 10%, IgG3 about 5%, and IgG4 may be undetectable in up to 20% of healthy persons. Additionally, serum levels are age related. It has been difficult to establish a link between IgG subclass deficiencies and any consistent pattern of infections. IgG replacement should be reserved for patients with defects in specific antibody production and recurrent infections, which is rarely seen in patients with selective IgA or IgG subclass deficiencies.
Treatment of Hypogammaglobulinemia
The mainstay of therapy for hypogammaglobulinemia is replacement with IgG, but appropriate management of infections is also important. Curative therapy with bone marrow transplantation (BMT) has been successful in patients with XL HIGM syndrome. Replacement IgG is usually given by intravenous infusions at a dose of 400–600 mg/kg every 3–4 weeks to maintain trough serum IgG levels above 500–800 mg/dL (a higher trough level is targeted for patients with established pulmonary disease). Subcutaneous replacement is available, but it requires more frequent injections and may limit maximum dosing. The aim of treatment is to prevent future infections and minimize any progression of chronic lung disease (bronchitis or bronchiectasis). Despite the passive immunity provided by replacement IgG, infections remain a persistent risk for affected patients. The prognosis also depends on timely and appropriate antibiotic therapy. Typical infecting organisms include encapsulated bacteria, but Ureaplasma and Mycoplasma species must also be considered. Infusions are generally well tolerated, with most reactions being mild, including headache, back and limb pain, anxiety, and chest tightness. Rare systemic reactions can occur, including tachycardia, shivering, fever, and, in severe cases, anaphylactoid shock. These adverse symptoms can be prevented by pretreatment with corticosteroids, antihistamines, and nonsteroidal anti-inflammatory drugs. For patients with congenital hypogammaglobulinemia, replacement therapy is currently lifelong. As a precautionary measure, patients with agammaglobulinemia or hypogammaglobulinemia should not receive live vaccines, but nonlive vaccines may be beneficial, particularly in patients with CVID.
SEVERE COMBINED IMMUNODEFICIENCY DISEASES
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


