Immune- and Inflammatory-Mediated Central Nervous System Syndromes
Mark P. Gorman
The central role of the immune system in several pediatric central nervous system (CNS) disorders has been increasingly appreciated in recent years. Although relatively rare as individual diseases, collectively they constitute a sizable proportion of pediatric neurology practice. The acute, severe symptoms associated with these disorders usually lead to inpatient hospitalization. Due to the broad differential diagnoses associated with these conditions, multiple other pediatric subspecialists, such as infectious disease and rheumatology physicians, are often asked to evaluate affected patients. Last, because of the potential long-term sequelae of both the monophasic and recurrent immune-mediated CNS disorders, all aspects of a patient’s medical and psychosocial care can be affected. Thus, these disorders have relevance to general pediatricians and pediatric subspecialists in both the inpatient and outpatient settings.
CNS IMMUNE-MEDIATED DEMYELINATING DISORDERS
Demyelinating disorders comprise the largest subgroup within CNS immune-mediated disorders. Myelin is composed of a lipid bilayer of cholesterol, phospholipids, and glycolipids along with membrane-associated proteins, such as proteolipid protein and myelin basic protein.1,2 In the CNS, oligodendrocytes produce myelin, which surrounds axons with periodic interruptions at nodes of Ranvier. The main functions of myelin are to speed the conduction of action potentials along axons and to support the development and maintenance of axons.
Myelin can be injured by many different mechanisms, such as hypoxia, metabolic derangements, and toxic insults. The disorders discussed in this chapter are considered to have an autoimmune etiology, with loss of tolerance resulting in an aberrant, self-reactive inflammatory process directed against CNS myelin.
Demyelination leads to slowing or blockade of action potential propagation, with resulting symptoms referable to the affected CNS areas. Although remyelination can occur in the CNS, the thickness of the original myelin sheath is never reachieved.3 Demyelination can also lead to secondary axonal loss, which leads to permanent disability.4
FIRST ATTACK OF DEMYELINATION
 DEFINITIONS, TERMINOLOGY, AND CLASSIFICATION
DEFINITIONS, TERMINOLOGY, AND CLASSIFICATION
In the past, variable terminology and lack of consistent definitions in the literature hampered our understanding of pediatric CNS demyelinating disorders. In April 2007, diagnostic definitions were proposed by the International Pediatric Multiple Sclerosis (MS) Study Group.5 Although they have not yet been prospectively validated, these definitions provide a very useful framework both for clinical and research purposes.
With a first presentation of a CNS demyelinating disorder, determining whether mental status changes are present or absent serves as the initial step in classification (Fig. 556-1). When present and accompanied by multifocal symptoms, the appropriate diagnosis is acute disseminated encephalomyelitis (ADEM), which can be confirmed with magnetic resonance imaging (MRI). When the patient’s mental status is normal, first-time acute demyelinating events are collectively referred to as clinically isolated syndromes (CIS). Clinically isolated syndromes can be subdivided based on whether the symptoms and signs are focal or multifocal. Common locations for focal CIS include the optic nerve (optic neuritis), spinal cord (transverse myelitis), brain stem, and cerebellum. If multiple CNS locations are involved simultaneously but the mental status is normal, the appropriate diagnosis is a polysymptomatic CIS.
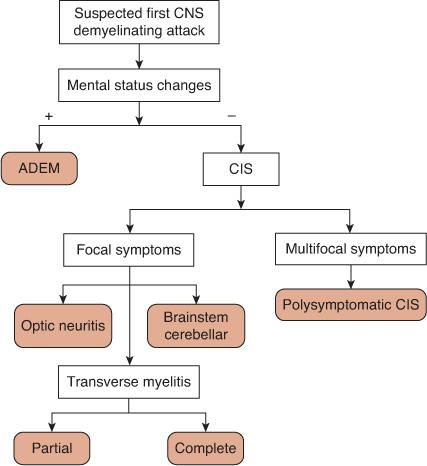
FIGURE 556-1. Algorithm for the classification of a first central nervous system (CNS) demyelinating attack. ADEM, acute disseminated encephalomyelitis, CIS, clinically isolated syndrome.
A patient with a CIS may have evidence of demyelination on MRI in numerous CNS sites without accompanying clinical symptoms and signs. Although such an MRI finding is important for the patient’s prognosis for the development of MS, it does not alter the diagnosis, which is based on clinical findings. For example, a patient with optic neuritis, normal mental status, and multiple, asymptomatic brain and spine MRI lesions should be classified as having optic neuritis, not ADEM, and is at high risk to develop MS.
Demyelination of the spinal cord (myelitis) can be partial with mild, asymmetric motor and sensory symptoms, or complete with severe, symmetric motor, sensory, and autonomic symptoms. Partial myelitis is a type of CIS and carries a high risk for the development of MS, and the complete form poses little risk of recurrence in the pediatric population. The term acute transverse myelitis (ATM) should be reserved for the complete form. Definitions of ATM have varied significantly in the literature. In 2002, the Transverse Myelitis Consortium Working Group published useful diagnostic criteria.6
 GENERAL ASPECTS OF THE DIFFERENTIAL DIAGNOSIS
GENERAL ASPECTS OF THE DIFFERENTIAL DIAGNOSIS
Although the differential diagnosis varies somewhat based on the site of CNS demyelination, general principles can be applied to all of the disorders. There are many conditions that can mimic and be mimicked by pediatric CNS demyelinating disorders. In addition, as there are no absolutely definitive diagnostic tests for CNS demyelinating disorders, a broad differential diagnosis must be considered. A complete discussion of the entire differential diagnosis is beyond the scope of this chapter but it has recently been reviewed.7 In common practice, one will obtain laboratory tests including erythrocyte sedimentation rate, C reactive protein, serum Lyme titers, antinuclear antibody, and vitamin B12 level in all patients with CNS demyelinating disorders, with additional testing if necessary as guided by the history and examination. In general, the differential diagnosis and subsequent workup should be expanded in patients with younger onset, progressive decline in function, poor response to immunomodulation, and extra-CNS organ system involvement. The main disease categories that should be considered are infectious diseases, rheumatological disorders, metabolic disorders, and neoplastic conditions.
Regarding infectious diseases, microorganisms can directly infect the CNS (Fig. 556-2A) or can infect peripheral organs and secondarily trigger an autoimmune response. If there is any clinical concern for CNS infection in a patient with suspected CNS demyelination, cerebrospinal fluid (CSF) should be examined for common infections, such as enterovirus, and serious, treatable infections, such as herpes simplex virus, with polymerase chain reaction testing. Additional infectious disease testing can be tailored based on the season of the year and the patient’s geographic region, exposures, and immunocompetence, with strong consideration given to testing for Borrelia burgdoferi, Mycoplasma pneumoniae, and Epstein-Barr virus.
Many rheumatological disorders such as systemic lupus erythematosus (SLE) have been associated with CNS complications, which may be partially mediated by inflammatory demyelination (Fig. 556-2B).  Primary and secondary CNS vasculitis can also mimic demyelination and should be considered, particularly when headaches are a prominent symptom, symptoms recur during steroid withdrawal, or MRI reveals significant cortical involvement (Fig. 556-2C).
Primary and secondary CNS vasculitis can also mimic demyelination and should be considered, particularly when headaches are a prominent symptom, symptoms recur during steroid withdrawal, or MRI reveals significant cortical involvement (Fig. 556-2C).
Leukodystrophies are genetic and metabolic disorders that preferentially affect the white matter and therefore mimic CNS demyelination. In patients with a subacute to chronic course, progressive degeneration, and symmetric white matter involvement on MRI, metabolic testing should be considered, with specific tests based on the patient’s age, presentation, and MRI findings (Fig. 556-2D). These disorders are discussed in Chapter 576.
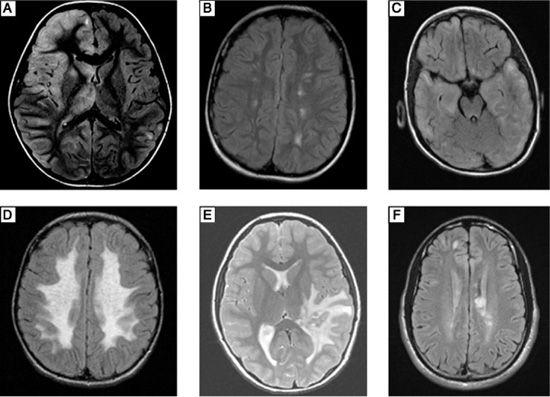
FIGURE 556-2. A: Axial fluid attenuated inversion recovery (FLAIR) magnetic resonance image (MRI) with hyperintense signal mainly in the right frontal cortex and deep gray matter of a 6-year-old boy who presented with fever, left-sided weakness and left-sided focal motor seizures. Cerebrospinal fluid polymerase chain reaction testing was positive for Epstein-Barr virus. B: Axial FLAIR MRI showing discrete hyperintensities in the subcortical white matter bilaterally in a 12-year-old girl who presented with chorea and hematuria. Workup revealed positive antinuclear antibody, and she was diagnosed with systemic lupus erythematosus (SLE). C: Axial FLAIR MRI with bilateral hyperintense signal in the subcortical white matter and cortex diffusely in a 5-year-old boy who presented with seizures and encephalopathy. Brain biopsy was consistent with primary CNS vasculitis. D: Axial FLAIR MRI showing bilateral symmetric hyperintense signal involving all of the cerebral white matter with sparing of the immediate subcortical region in a 7-year-old boy who presented with a 2-year history of progressive cognitive decline followed by ataxia. Biochemical and genetic testing was consistent with metachromatic leukodystrophy. E: Axial T2-weighted MRI showing a large heterogeneous lesion in the deep white matter of the left temporal-parietal region with extensive surrounding edema mimicking a tumor in an 8-year-old girl who presented with headaches, diplopia, and seizures. Biopsy of the lesion revealed perivascular lymphocytic and monocytic infiltration with patchy demyelination. F: Axial FLAIR MRI showing discrete, ovoid hyperintense lesions involving the periventricular more than the subcortical white matter in a 17-year-old boy who presented with right-sided numbness. His diagnostic workup and subsequent course was consistent with relapsing-remitting multiple sclerosis.
Neoplasms can be confused with demyelination, particularly when there is a large, focal inflammatory lesion (ie, tumefactive demyelination) (Fig. 556-2E). MRI of the entire neuroaxis to look for multifocal involvement and cytological examination of the CSF should be obtained in such circumstances. Rarely, biopsy of the lesion is needed to make a definitive diagnosis.
 GENERAL ASPECTS OF ACUTE TREATMENT
GENERAL ASPECTS OF ACUTE TREATMENT
The treatment of pediatric CNS demyelinating disorders can be generally divided into acute and prophylactic phases. Acutely, attacks of demyelination are treated similarly, regardless of etiology (Fig. 556-3). High-dose intravenous (IV) corticosteroids are the first-line treatment. 
Although not formally tested in pediatric patients, high-dose IV corticosteroids are used for the initial treatment of acute attacks of demyelination. The most commonly used protocol consists of methylprednisolone 20 to 30 mg/kg/dose (maximum 1 gram) intravenously once a day for 3 to 5 days. During use of this steroid, pulse, vital signs, especially blood pressure, and glucose control should be monitored. Ulcer prophylaxis should be used. Common adverse side effects include insomnia and mood changes.
The use of oral steroid tapers following intravenous treatment is controversial. Patients who have complete or nearly complete symptom resolution may not need tapers, but those with incomplete recovery may benefit from a 2- to 3-week taper. Due to the adverse effects of chronic corticosteroid use and the availability of other immunomodulatory agents, prolonged courses of oral steroids for the treatment of CNS demyelinating disorders is not recommended.
If significant functional disability remains after several days of observation following high-dose steroid treatment, additional options include intravenous immunoglobulin (IVIg) and plasma exchange. Based on case reports, IVIg 0.4 grams/kg for 5 days or 1 gram/kg for 2 days can be used as second-line treatment.11,12 The mechanism of action of IVIg is uncertain, but likely involves decreased production of autoantibodies.13
If IVIg is ineffective, plasma exchange administered every other day for a total of 5 exchanges can be considered. This regimen has been used successfully in 1 case series of 6 pediatric patients with ADEM who did not respond adequately to steroids and IVIg.14
ACUTE DISSEMINATED ENCEPHALOMYELITIS (ADEM)
 EPIDEMIOLOGY
EPIDEMIOLOGY
Acute disseminated encephalomyelitis (ADEM) is defined as an acute or subacute inflammatory demyelinating event affecting multifocal areas of the CNS with multiple accompanying symptoms, which must include encephalopathy.5 The incidence of ADEM is approximately 4 cases per 1 million persons under the age 20 per year.18 It is more common in children than adolescents and adults, with a mean age of onset between 5 and 8 years of age.
 PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
Although the pathophysiology of ADEM is likely autoimmune mediated, the precise mechanisms remain uncertain. Indirect evidence implicating the immune system includes the frequent association with a preceding viral infection or, less commonly, a vaccination, as well as the apparent effectiveness of immunomodulation. The former point suggests that molecular mimicry, whereby epitopes on microorganisms mimic those found in CNS myelin and thus provoke an autoimmune response, may play a role in the pathophysiology of ADEM. As most of the infections reported to act as triggers for ADEM are very common, additional host susceptibility factors must be present in patients with ADEM but are largely undefined.
Both B and T lymphocytes appear to play a role in ADEM. Regarding B cells, autoantibodies to myelin oligodendrocyte glycoprotein were found in 20% of patients with ADEM.20 Antimyelin autoreactive T cells have also been found in the serum of a small group of patients with ADEM.21 Additional work is needed to clarify the immune and genetic mechanisms of ADEM.
Although biopsies are rarely needed to establish the diagnosis of ADEM, those that are performed provide more direct evidence of the role of the immune system. Biopsy specimens typically show perivenous infiltrates of lymphocytes and macrophages with accompanying demyelination and relative axonal sparing (Fig. 556-4).
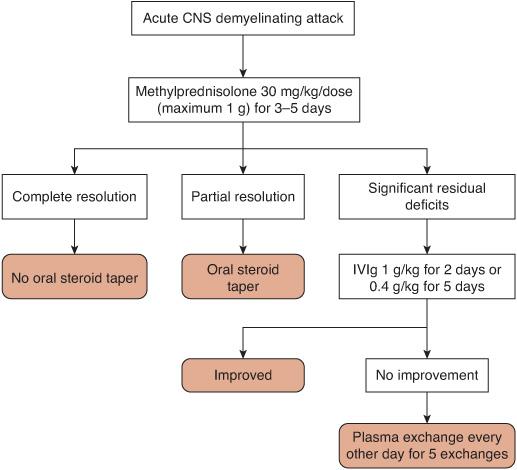
FIGURE 556-3. Algorithm for the treatment of an acute central nervous system (CNS) demyelinating attack. IVIg, intravenous immunoglobulin.
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Children with ADEM present with a combination of nonspecific symptoms and signs suggestive of meningoencephalitis, as well as focal neurologic deficits. Approximately 70% of cases follow a viral illness or vaccination, usually occurring 7 to 14 days prior to the onset of neurological symptoms. The most common associated infection is a nonspecific upper respiratory tract infection. Pooled data from nine case series totaling 411 patients yield the following estimates for presenting symptoms and signs: motor (60%), fever (50%), headache (40%), vomiting (40%), ataxia (40%), cranial nerve deficits (40%), seizures (25%), and vision loss (15%).18,22-29 Thus, a typical clinical scenario is that of a previously healthy child who develops a nonspecific infection, fully recovers, and then develops acute encephalopathy and multifocal neurological symptoms and signs.
 DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
As suggested by these symptoms, the presentation of ADEM can be indistinguishable from meningoencephalitis. Thus, direct CNS infection should be ruled out with lumbar puncture and appropriate studies in all patients with suspected ADEM. Cerebrospinal fluid (CSF) analysis is abnormal in approximately 60% of patients with ADEM, with the most common findings being lymphocytic pleocytosis or elevated protein concentration, or both.18,22-29 Thus, basic CSF studies may not distinguish ADEM from encephalitis, and testing for specific causes of viral encephalitis should be performed. Additional testing should be guided by clues in the history, examination, or neuroimaging as described above.
 DIAGNOSTIC EVALUATION
DIAGNOSTIC EVALUATION
All patients with acute disseminated encephalomyelitis (ADEM) should undergo contrastenhanced MRI of the brain and spine. Although patients with ADEM may initially require computerized tomography (CT) of the head to rule out other emergent conditions, CT has a poor sensitivity of approximately 40% when compared to MRI in patients with ADEM.18,22-29 MRI typically shows bilateral, asymmetric, multifocal hyperintense lesions on T2 and fluid-attenuated inversion recovery (FLAIR) sequences. The lesions predominantly affect the cerebral white matter, more so in subcortical rather than periventricular regions. However, cortical, deep grey nuclei, brain stem, cerebellar, and spinal cord lesions are also seen (Fig. 556-5). Lesions tend to be large with ill-defined borders and lack contrast enhancement.
 TREATMENT
TREATMENT
The initial treatment of ADEM is similar to that used for all acute attacks of CNS demyelination (see above). In addition to immunomodulation, supportive care during the acute attack may include airway management in patients with depressed mental status, anticonvulsants in patients with seizures, and pain management. As ADEM is a terrifying event for patients and their families, psychosocial support should also be given. By definition, ADEM is a monophasic disorder and does not require chronic immunomodulation. However, patients with ADEM are at risk for long-term sequelae (see below) and therefore should be followed by a pediatric neurologist with treatment of cognitive deficits, physical handicaps, and epilepsy, as appropriate.
 PROGNOSIS
PROGNOSIS
Approximately 80% of patients with ADEM make a full recovery.18,22-29 Most show significant improvement prior to hospital discharge, with the remainder of the recovery occurring gradually over several months. However, some patients are left with motor (10–15%), cognitive or behavioral (10%), epileptic (5–10%), or visual (5%) sequelae.18,22-29 Cognitive sequelae may be more common than previously appreciated, as suggested by 2 recent studies that demonstrated a high percentage of cognitive deficits with formal neuropsychological testing, particularly in patients with the onset of ADEM at less than 6 years of age.30,31 Thus, children with ADEM should be followed closely for declines in cognition or school performance following the acute illness.
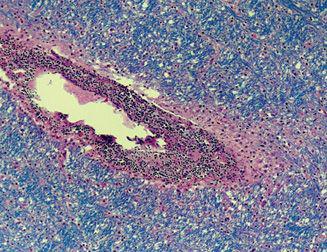
FIGURE 556-4. Autopsy specimen of the subcortical white matter from an 18-year-old boy who presented with lethargy, seizures, and ataxia following a viral infection. Despite treatment, the patient developed brain herniation and died. The specimen shows intense perivenular mononuclear cell infiltration (round, purple cells) with associated demyelination indicated by absence of Luxol fast blue staining in the perivenular white matter. The remainder of the white matter is relatively spared. (Source: Courtesy of Hart Lidov, MD, Department of Pathology, Children’s Hospital, Boston.)
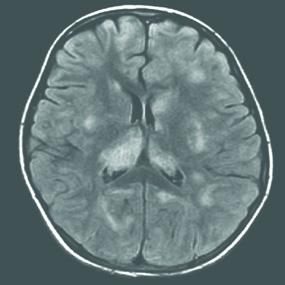
FIGURE 556-5. Axial fluid attenuated inversion recovery magnetic resonance image (MRI) showing multifocal, large, ill-defined hyperintense lesions in the subcortical white matter, basal ganglia, and cortex in a 9-year-old girl with acute disseminated encephalomyelitis (ADEM) who presented with fever, headaches, encephalopathy, and weakness following a viral infection. Following steroid treatment, the symptoms and MRI findings resolved.
Approximately 80% of patients with ADEM experience a single event without recurrences.18,22-29 Based on the expert opinion of the International Pediatric MS Study Group, symptoms occurring within 3 months of the initial onset of ADEM can be considered part of the initial event.5
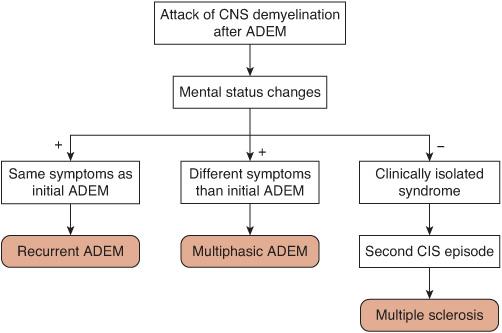
The reemergence of the initial symptoms after 3 months has been designated recurrent ADEM. If the symptoms are different than the initial presentation, the term multiphasic ADEM can be used5 (Fig. 556-6). Both recurrent and multiphasic ADEM require that the relapses be accompanied by mental status changes. Multiphasic ADEM is less common than recurrent ADEM, occurring in less than 5% of patients overall.18,22-29 Given the relative rarity of recurrent and multiphasic ADEM, a broad differential diagnosis should be reconsidered before making these diagnoses. Patients with recurrent or multiphasic ADEM do not generally require chronic immunomodulation, but it could be considered for unusual patients with frequent or severe attacks. If mental status changes are not present during the second event following ADEM, it will most likely represent a CIS-type event (ie, optic neuritis). 
The percentage of patients with an initial diagnosis of ADEM who later develop MS is uncertain. Estimates of this risk have ranged from 0%26 to as high as 18%32 in the existing literature, with a pooled average of approximately 10%.18,22-29 Although there are no highly predictive risk factors, optic nerve involvement with ADEM, family history of CNS demyelination, and MRI features suggestive of MS may increase the risk of developing MS following ADEM.32
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


