Disorders of varied biological behavior
Malignant disorders
Dendritic cell related
• Langerhans cell histiocytosis
• Secondary dendritic cell processes
• Juvenile xanthogranuloma and related disorders
• Solitary histiocytomas of various dendritic cell phenotypes
Macrophage-related
1. Hemophagocytic syndromes
• Primary hemophagocytic lymphohistiocytosis: Hemophagocytic syndromes( familial and sporadic)
• Secondary hemophagocytic syndromes:
Infection-associated
Malignancy-associated
Other
2. Rosai–Dorfman disease (sinus histiocytosis with massive lymphadenopathy)
3. Solitary histiocytoma with macrophage phenotype
Monocyte-related
1. Leukemias (FAB and revised FAB classifications)
• Monocytic leukemia M5A and B
• Acute myelomonocytic leukemia M4
• Chronic myelomonocytic leukemia
2. Extramedullary monocytic tumor or sarcoma
Dendritic cell-related
• Histiocytic sarcoma (localized or disseminated)
Specify phenotype, follicular dendritic cell, interdigitating dendritic cell, etc.
Macrophage-related
• Histiocytic sarcoma (localized or disseminated)
Dendritic Cell Lineage
Langerhans cell histiocytosis (LCH), previously known as Histiocytosis X, is a condition characterized by a clonal proliferation of CD1a+ LC. LCH is the type of histiocytosis observed with higher frequency [5, 11]. Clinical presentation and prognosis vary depending on the site and number of organs affected. Organs most often involved are bones, skin, lymph nodes, and parenchymal organs (liver, spleen, lungs, and bone marrow) [12–15]. Children with LCH may present with single-system (SS) or multisystem (MS) disease and have diverse outcomes: it may resolve spontaneously, respond well to the therapy, become a chronic recidivating condition, or have a rapidly fatal result [12].
Epidemiology
LCH is rare in children.
LCH affects 5–6 million children per year between the ages of 1 and 15.
Incidence in children <1 year is around 15 million per year.
LCH is considered a rare disease. In children under the age of 15 it has been estimated to affect 2.2–8.9 per million every year [13, 16]. Since 30–50 % of cases present in patients older than 15 years, the actual incidence for pediatric age is likely higher, probably around 11–12 per million/year [5]. Incidence rates range from 1.3 per million per year in children age <1 year to 2.0 per million per year in the 10–14-year age group [17]. Several studies suggest that there is a slightly higher prevalence in males 2.2–1.1/1 [18, 19]. In general, median age of diagnosis has been reported between 1 and 4 years of age [13, 14]. There is no ethnic association; however it has been reported to be very rare in blacks [19].
Etiology
The precise etiology of LCH is unknown [20]. The immune mechanisms underlying LCH are still not fully understood, although important progress has been recently made. Controversy remains whether LCH is a reactive or a neoplastic disorder. Some of the studies that support the latter have demonstrated that LCs from non-pulmonary lesions are monoclonal based on a pattern of highly skewed X-chromosome inactivation [21]. There is also evidence of cell-cycle deregulation within lesions, and the presence of telomere shortening of LCH cells [22]. Its association with lymphoma and acute lymphoblastic leukemia also upholds this theory [21]. Studies in twins and familial presentation have also suggested a possible genetic predisposition [22]. Another theory states that LCH is a reactive disease due to an immature deregulation between LCs and T lymphocytes. Viruses including human herpes virus type 6, cytomegalovirus, and Epstein–Barr virus have been implicated as triggers for this process; however, there is not enough evidence to support the hypothesis that LCH results from a viral trigger [22]. Smoking has also been associated with LCH in adults. It has been shown that cigarette smoke increases the number of LCs in the bronchial epithelium and might be one of the precipitants for pulmonary LCH [20]. More recently, Badalian-Very et al. demonstrated that 35/61 cases of LCH (57 %) exhibit somatic activating mutations in the proto-oncogene BRAF-V600E1 [23]. This has been previously observed in premalignant cutaneous nevi, melanoma, papillary thyroid cancer, colorectal and lung cancer, low-grade ovarian carcinoma, and pediatric low-grade glioma [21]. On the basis of LCH cell clonality and the presence of activating somatic BRAF mutations in most patient samples, some authors consider that LCH needs to be considered as a neoplastic disease [24].
Classification
Historically, different variants of LCH were described: eosinophilic granuloma, Letterer–Siwe disease, Hand–Schüller–Christian syndrome, Hashimoto–Pritzker syndrome, self-healing histiocytosis, pure cutaneous histiocytosis, LC granulomatosis, type II Histiocytosis, and non-lipid reticuloendotheliosis. Later, all these were grouped under the name of Histiocytosis X by Lichtenstein in 1953 [25].
Currently, LCH is more commonly classified according to organ system involvement and then subdivided into single or multisite involvement [13]. The Histiocyte Society’s treatment protocol for LCH categorizes patients as those with single-system disease (involving a single site), those with single-system disease affecting multiple sites (multifocal), and those with multisystem disease [10].
Clinical Presentation
Clinical manifestations vary widely depending on organs/systems affected.
LCH is classified into single-system disease or multisystemic disease.
Most common organ affected is the bone.
Skin is the second most commonly affected organ.
Skin manifestations are polymorphous: red papules, vesicles, hemorrhagic, scaly, and seborrheic-like eruptions have been described.
Scalp and folds are more commonly affected.
Single-System Disease
Isolated organ involvement is termed single-system disease (SS-LCH). The skeletal system is the most commonly involved location in LCHs. Lesions may manifest as painless, isolated bone lesions or as multiple lesions accompanied by severe dysfunction and deformities [20]. In children the skull and femur are the most common sites affected [26]. The term eosinophilic granuloma was previously used to describe the chronic localized form of LCH affecting bone [8]. Up to 40 % of lesions affecting the skull may associate diabetes insipidus due to pituitary infiltration [26]. Historically, the triad consisting of bone disease, diabetes insipidus, and exophthalmos in a child with chronic, progressive multifocal involvement was called Hand–Schüller–Christian disease [8, 19].
The second most commonly affected organ is the skin [15, 16, 18] Skin lesions are typically the first manifestation of LCHs in patients under 1 year of age [27, 28]. LCH can present with a wide range of cutaneous findings (Figs. 23.1, 23.2, 23.3, and 23.4). The three main types include: (1) infiltrating lesions (nodules and/or plaques that may ulcerate), (2) extensive papules and plaques with scale or crusts, and (3) seborrheic dermatitis-like or eczematous-like lesions [29]. Other lesions can be purpuric, and present with necrosis. The spectrum of color found on the cutaneous manifestations goes from hypopigmented, yellowish, red-brown to hyperpigmented lesions and this can vary according to the underlying Fitzpatrick skin type [30] (Table 23.2).
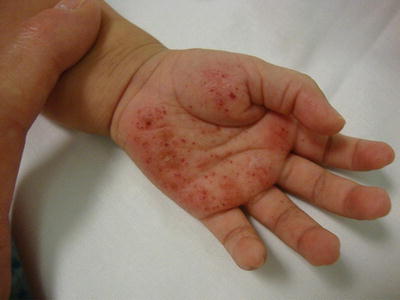
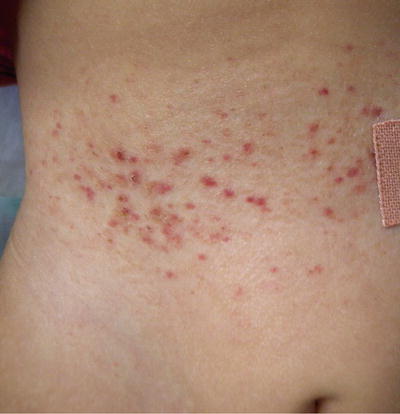
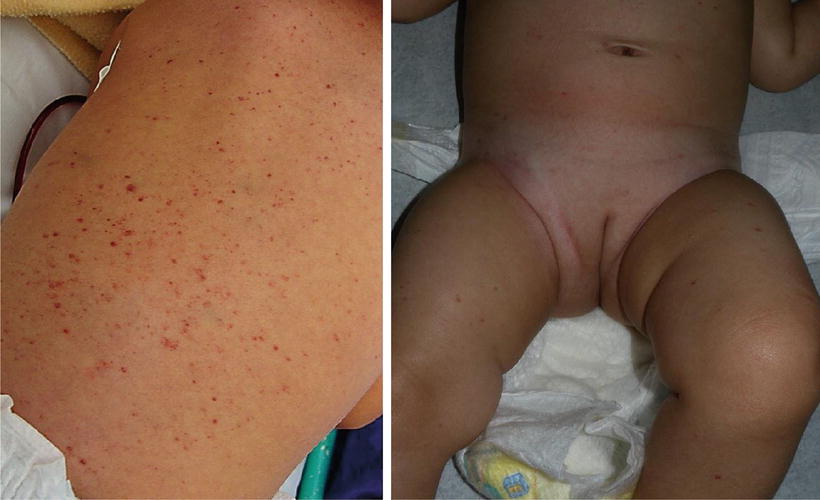
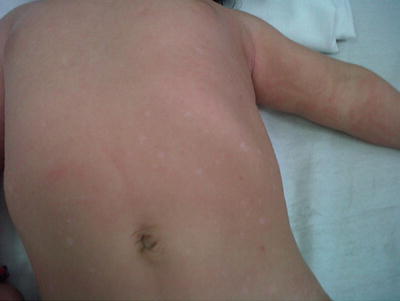
Fig. 23.1
Infiltrated erythematous papules with purpura and crusting on the palm and abdomen in patient with LCH. Courtesy Dr. Duran-McKinster
Fig. 23.2
Infiltrated erythematous papules with purpura and crusting on the palm and abdomen in patient with LCH. Courtesy Dr. Duran-McKinster
Fig. 23.3
(a) Multiple infiltrated erythematous papules with purpura on the back of patient with LCH. Courtesy Dr. Duran-McKinster, (b) Scattered erythematous papules on an infant with LCH
Fig. 23.4
Hypopigmented macules on the abdomen of a child with LCH. Courtesy Dr. Duran-McKinster
Table 23.2
Skin lesions described in children with Langerhans cell histiocytosis
• Infiltrating nodules/plaques (may be ulcerated) |
• Flesh-colored to yellow or red-brown papules covered with scales/crust |
• Hypopigmented papules |
• Papular xanthomas-like |
• Seborrheic dermatitis-like lesions (scalp) |
• Eczematous dermatitis-like lesions |
• Bilateral auricular discharge |
• Perianal lesion (vegetative/ulcerated) |
• Purpuric/petechiae and necrotic lesions |
• Intertriginous lesions (diaper, behind the ears, axillae) |
• Molluscum contagiosum-like lesion |
• Blueberry Muffin lesion, vesicles/bullae lesions(congenital) |
• Darier’s disease-like and urticarial lesion (less often described) |
• Nodules and mucosal ulcerations (altered dentition) |
• Nail changes include dystrophy, paronychia, and bed nail bleeding |
The most common site of skin involvement is the scalp, followed by the trunk and groin; however, several case reports also include perianal, genital, limb, and facial [11, 28, 30–33]. Mucosal and nail disease are characterized by nodules and/or ulcerations, but disruption of tooth eruption, nail dystrophy, paronychia, and hemorrhagic bed nails can also be seen [34].
Among skin-only LCH, congenital self-healing reticulohistiocytosis (CSHRH) deserves special consideration in children. Previously known as Hashimoto–Pritzker disease, this condition usually presents at birth in an otherwise healthy baby and tends to resolve spontaneously during the first months of life. Progression of CSHRH to a multisystem disease has been reported, but some authors consider that this may represent two different entities, since CSHRH and the progressing form of skin-only LCH are indistinguishable clinically and immunohistochemically. Multisystem LCH in the neonatal period has poor prognosis and has been associated with high mortality rates. It has been reported that half of patients initially diagnosed as a skin-only condition actually had multisystem disease [35, 36]. Skin manifestations during the neonatal period do not differ from other age groups: red-brown papules, nodules, seborrheic-like lesions, vesicles, pustules, and hemorrhagic lesions are commonly described, and particular to this age group a “blueberry muffin baby” has also been reported [35].
Multisystemic Disease
Multisystemic disease is diagnosed when two or more systems are affected [18]. MS-LCH is subclassified as low risk or high risk depending if there is or no involvement of lymph nodes, bone marrow, spleen, lungs, or liver [37]. Bone involvement including facial bones, sinuses, maxilla, anterior or middle cranial fossa, with intracranial tumor extension, is also considered high risk [18]. Virtually, almost every organ or organ system can be affected by LCH. Multisystemic disease may or may not be characterized by organ dysfunction [13].
Multisystemic disease, formerly Letterer–Siwe disease, is the most aggressive form of LCH. Its frequency in relation to other LCH types varies from 3 to 75 % depending on the series reported [15, 16, 27, 28]. In children, MS disease tends to occurs more frequently before the age of 5 years with a peak incidence between 1 and 4 years [28]. Initial symptoms include anorexia, failure to thrive, and fever. Other manifestations depend on the specific organ affected. The most frequently reported include local pain or swelling (bone involvement), erythematous or weeping eczematoid rash affecting scalp, ear canals, abdomen, and intertriginous areas, including diaper, and otitis media with discharge, cough, tachypnea, hemoptysis, lymphadenopathy, and hepatosplenomegaly [26, 34]. Patients also have laboratory abnormalities like elevated sedimentation rate, thrombocytosis, leukopenia, anemia, and thrombocytopenia [16].
Diagnosis
Once the diagnosis of LCH is suspected, a full work-up (complete blood count with differential, blood chemistries including liver function tests, coagulation studies, urinalysis) and a biopsy from the organ affected or the most accessible lesion should be taken. Histological findings of LCH are distinctive. Lesions are characterized by an accumulation of mononuclear and multinucleated LC mixed with abundant mature eosinophils, as well as some neutrophils and small lymphocytes. LC have grooved, folded, indented, or lobulated vesicular nuclei. They are typically positive for immunohistochemical stains CD1a, langerin (CD207), S-100 protein, and CD68. The distinctive features of LCH are the Birbeck granules, a rod- or tennis-racket-shaped structure seen in electron microscopy; however, the latter is no longer recommended since it has been shown that the expression of Langerin fully correlates with the presence on electron microscopy of Birbeck granules [18, 19]. Additionally, skeletal radiographs (X-rays), abdominal ultrasound, and MRI are also necessary for diagnosing bone, liver, spleen, or central nervous system involvement.
Treatment
In SS-LCH disease lesions may resolve spontaneously; thus observation alone is acceptable.
Local treatment such with curettage and/or topical or intralesional steroids may be indicated in some cases.
In MS-LCH treatment includes vinblastine and predni-solone.
In high-risk patients with reactivation, prolonging initial chemotherapy has reduced mortality.
There are different treatment modalities for LCHs depending on the affectation or number of sites involved. Patients with unifocal or single disease may receive no treatment other than observation, since most patients recover spontaneously, or they can be given, in the case of bony lesion, local treatment such as curettage, intralesional steroid injections, or localized radiation. Oral indomethacin is another option for bone lesions due to its action as an analgesic and anti-inflammatory agent. For skin lesions topical steroids are the standard option [20]. Mustard 0.02 % applied topically and PUVA have also been reported to be effective to treat skin lesions of LCHs [22, 26].
Systemic therapy for multifocal or multisystemic disease is guided by hematology/oncology teams and includes chemotherapeutic protocols. Hematopoietic stem cell transplantation is also a current therapeutic option, with a moderate success, for adults and children if high-risk organ involvement is present, or they don’t respond to other therapeutic approaches [18, 38].
Prognosis
Patients with SS-LCH have an excellent prognosis, although sequelae have been reported. Bone sequelae develop in 3–42 % of patients [37] and include compression of vertebral bodies, orthopedic deformities, growth plate involvement, destruction of the orbit, tooth loss, and hearing impairment. Diabetes insipidus presents as a result of cranial bone involvement and may be accompanied by growth hormone deficiency [20]. This is most often seen in patients with multisystemic disease (until 40 %). In the skin, the most common cutaneous sequelae is scarring, (up to 30 %) and its extent and severity depend on the side of involvement [37].
The risk of SS-LCH disease progressing to MS-LCH disease is about 10 % [19]. For children with multisystemic disease and on recommended treatment an overall survival rate at 5 years of 94.4 % has been recently reported [22]. Children younger than 2 years at diagnosis have a higher risk of permanent consequences and increased morbidity and mortality due to the likelihood of developing multiorgan dysfunction [13, 27]. However, the mortality in these patients has been reduced to 10–20 % after the introduction of combined schemes of steroids and cytostatics [39, 40]. Disease recurrence in children varies depending on the series but has been reported up to 50 % in multifocal bone disease when patients are treated with single-agent chemotherapy, radiotherapy, or observation only [26].
Ongoing Research
The Histiocytosis Association Research Program’s primary objective is to identify and fund initiatives aimed at better understanding the physiopathology of LCH and finding more effective treatments and a potential cure. The following are some of the ongoing studies on LCH:
Analysis of Jagged2 Signaling in Langerhans Cell Histiocytosis
Caroline Hutter MD, PhD
Children’s Cancer Research Institute/St. Anna Kinderspital—Vienna, Austria
Research Infrastructure for Clinical and Translational Research in Langerhans Cell Histiocytosis (LCH)
Carlos Rodriguez-Galindo MD
Dana-Farber Cancer Institute—Boston, Massachusetts USA
Whole Exome Sequencing of Langerhans Cell Histiocytosis
Barrett Rollins MD, PhD; Astrid G.S. van Halteren PhD
Dana-Farber Cancer Institute—Boston, Massachusetts USA
Leiden University Medical Center—Leiden, The Netherlands
Histiocyte Society Clinical Trials Database System LCH-IV Treatment Protocol Study
Histiocyte Society Executive Board
LCH Study Reference Center—Vienna, Austria
Juvenile Xanthogranuloma and Related Disorders
Juvenile xanthogranuloma (JXG) and related disorders are a miscellaneous group of dendritic cell disorders that have a common denominator: the accumulation of histiocytes that do not meet the phenotypic criteria for the diagnosis of LCH, which are Birbeck granules, S-100 +, CD1a+ [41]. As proposed by Weitzman and Jaffe in 2005, the JXG family is comprised of different entities that can be divided into (1) cutaneous non-LCH such as benign cephalic histiocytosis, JXG, generalized eruptive histiocytoma (GEH), adult xanthogranuloma, and progressive nodular histiocytosis, (2) cutaneous with major systemic component like xanthoma disseminatum, and (3) systemic non-LCH or Erdheim–Chester disease [42, 43] (Tables 23.3 and 23.4).
Table 23.3
Juvenile xanthogranuloma and related disorders
Epidemiology | Clinical presentation | Treatment | |
|---|---|---|---|
BCH | Infancy <1 year | Multiple lesions Yellow to brown papules Face and neck | Self-resolving (8–48 months) |
JXG | Most common of non-LCH Infancy and childhood | Solitary to multiple lesions Yellow-brown papules or nodules Systemic involvement Variants: giant, mixed or plaque-like, clustered Face, neck, trunk | Self-resolving (1–6 years) Surgical removal |
GEH | Adults | Red-brown to red-blue or pale erythematous papules and/or nodules Trunk and proximal limbs | Self-resolving |
PNH | Young adults in their 3rd–4th decades | Several to hundred yellowish-brown papule/nodules Trunk, head, neck | Non self-resolving, surgical treatment |
XD | 3rd decade of life Males | Hundred of red-brown to yellow discrete papules and nodules Face and trunk and affects flexures and folds Mucosal involvement progressive forms with systemic involvement | Seldom self-resolving (5–28 years) Surgical excision, electrocoagulation, cryotherapy, radiation, antimetabolites, anticancer drugs |
ECD | Usually adults | Long bones, skin, retro-orbital tissues Systemic involvement Multiple yellow to red-brown papules/plaques Symmetric distribution | Corticosteroids, radiotherapy, chemotherapy, and immunotherapy |
ICH | No age predilection | Red to brown multiple papules/nodules distributed mainly over the trunk and extremities | Surgical removal, chemotherapy, 5 % fluorouracil, thalidomide, pravastatin, narrowband UVB |
Table 23.4
Macrophage-related disorders
Epidemiology | Clinical presentation | Treatment | |
|---|---|---|---|
HLH | Infancy and childhood | Genetic and acquired Fever (severe and persistent), bicytopenias, (platelets <100 × 109/L, neutrophils <109/L), hepatitis and splenomegaly Skin: purpuric, macular, papular, erythrodermic, or morbilliform eruptions | Combinations of immunosuppressive therapy and pro-apoptotic chemotherapy |
SHML | Children and young adults | Painless bilateral, massive cervical lymphadenopathy Skin: xanthoma-like, yellowish or reddish-brown papules/nodules Respiratory and digestive system involvement | Corticosteroids, chemotherapy, low-dose interferon, and radiation therapy |
MRH | Adults | Involvement of skin, joints (arthritis), and mucosa Solitary or multiple coalescing tender papule/nodular lesions Face, scalp, hands mainly dorsum, lateral fingers, and periungual | Nonsteroidal anti-inflammatory drugs (NSAIDs) corticosteroids, etanercept, methotrexate, and cyclophosphamide |
The JXG family has a broad clinical spectrum, ranging from solitary or multiple skin lesions to large deeply situated masses to widespread disease with systemic involvement. Histological appearance of all subtypes is very similar and often indistinguishable from one another. They present positivity to factor XIIIa, CD68, CD163, l fascin, CD 14, and are negative for CD1a and S-100 proteins [42, 44].
Epidemiology
Disorders belonging to the JXG family are considered uncommon, and their specific frequency is unknown. These disorders have been reported in children and adults of all races. JXG is considered by far the most common type of non-LCH [8].
Cutaneous Non-LCH
Benign cephalic histiocytosis (BCH) (Figures 23.5, 23.6, 23.7, and 23.8) is an uncommon presentation of non-LCH. After been described by Gianotti et al. in 1971, more than 50 cases in various races have been reported [45]. The literature does not provide any data on the frequency or geographic variation of BCH. BCH usually presents during the first year of life (mean age of 13.1–14.3 months) with 50 % occurring before the age of 6 months [45]. Most publications agree that there is no difference in gender, but a slight predominance in males has also been reported [46]. Spontaneous regression of the lesions typically begins within 8–48 months with an average of complete remission at 50 months [47, 48]. BCH presents as an asymptomatic eruption of yellow to red-brown papules. Most commonly it affects the face and neck, but is also seen in upper extremities and trunk [45, 49, 50].
Lesions start as yellow to brown maculae that progress to papules ranging in diameter between 1 and 10 mm (Figs. 23.5 and 23.6). Patients can present with 1 to over 100 lesions and these lesions can coalesce to form a reticulate pattern [46]. Resolving lesions may leave behind hyperpigmentation or atrophy [51] (Fig. 23.7). There is no mucous membrane or visceral involvement; however ocular lesions have been reported [47].
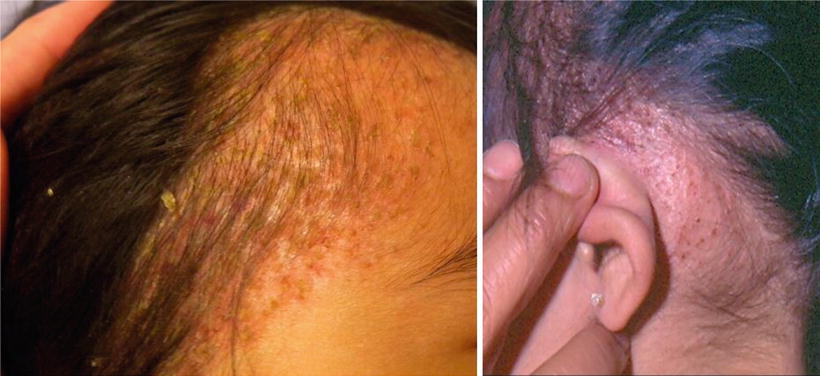
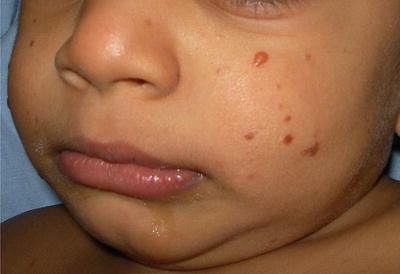
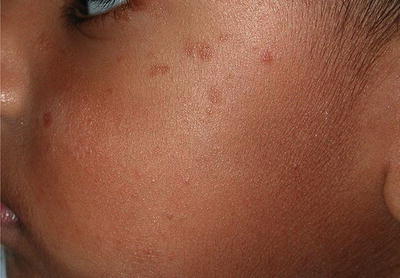
Fig. 23.5
(a) Seborrheic dermatitis-like lesions in patient with BCH. (b) Retroauricular infiltrated papules in patient with BCH (courtesy Dr. Duran-McKinster)
Fig. 23.6
Dark brown papules in an infant with BCH
Fig. 23.7
Residual mild atrophy and hyperpigmentation in a child with BCH
Extensive investigations for systemic involvement are not required [52]. Some reports mention a possible association between BCH with diabetes insipidus and insulin-dependent diabetes mellitus [53], but other authors consider these findings incidental [45].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


