Anemia
Physiologic anemia is well recognized during pregnancy, resulting from an expansion of maternal plasma volume that occurs to a greater degree than the pregnancy-induced expansion of red cell mass. Maternal anemia can also be present or develop during pregnancy because of deficiencies of essential hematinics such as iron, vitamin B12 and folate. Such pathologic anemia can adversely affect fetal growth and development and also increases the risk of maternal morbidity and mortality mainly at delivery if postpartum hemorrhage occurs. Severe anemia is common in women from resource-poor countries with an estimated 75% of women having anemia. However, even in industrialized countries, anemia mainly due to iron deficiency is common, especially in less advantaged groups. The increased hematinic requirements during normal pregnancy can further compound deficiency states and anemia in women. Correction of anemia in the antenatal period may improve maternal and fetal outcomes. The WHO definition of anemia in pregnancy is a hemoglobin of less than 110 g/L (11 g/dL) but many laboratories will define their own pregnancy normal ranges that may be as low as 100 g/L (10 g/dL).
Iron and iron deficiency
Iron is an essential component of hemoglobin and deficiency of iron will ultimately lead to anemia. In the absence of bleeding, only a small amount of iron is lost daily, mainly through shedding of intestinal mucosal cells so that iron deficiency is uncommon in men who have a diet with high iron bio-availability. In women menstrual blood loss places a continual demand on iron stores so that iron deficiency is much more common if dietary iron intake is insufficient to offset losses due to menstruation, especially in women with menorrhagia. Studies suggest that one in 10 women of reproductive age in developed countries is iron deficient, with iron deficiency anemia (IDA) present in 2–5% [1]. Adolescent women have a higher prevalence of iron deficiency and IDA likely to reflect iron requirements for growth during this period coupled with less reliable dietary intake of iron.
Iron requirements in pregnancy
The additional iron requirements for pregnancy are estimated to be around 1000 mg, with 45% of the total required for expansion of the maternal red blood cell mass, about one-third for fetal and placental growth and the remainder made up of obligatory basal intestinal losses and maternal blood loss at delivery. The increased demand is not uniform throughout pregnancy, with the greatest fetal demand for iron occurring in the last trimester of pregnancy. Iron requirements during the first trimester of pregnancy are estimated to be 0.8 mg/day of elemental iron, 4–6 mg/day from the second trimester onwards increasing to as high as 8–10 mg/day in the last 6 weeks of pregnancy. The term elemental iron refers to the amount of absorbable iron in any supplement and varies with formulation of iron used (see below). The predominant transfer of iron to the fetus occurs in the third trimester. There is active placental transport of iron via specific placental iron receptors ensuring continued supply of iron for fetal development [2].
For women who eat a diet with high iron bio-availability, pre-pregnancy iron stores of around 300 mg are thought to be sufficient to prevent iron deficiency developing during pregnancy. Iron stores are dependent on factors such as dietary intake, degree of menstrual blood loss, number of previous pregnancies and lactation. Iron deficiency and IDA are more common in adolescents and women with a higher parity. Median iron stores in women from industrialized countries vary between 150 mg and 300 mg but an alarming 20% of women enter pregnancy with no iron stores. In less developed countries with low bio-availability iron diets and other factors that contribute to anemia, a very high proportion of women can be expected to enter pregnancy with low or no iron stores.
Iron deficiency and iron deficiency anemia: prevention and treatment
Dietary intervention
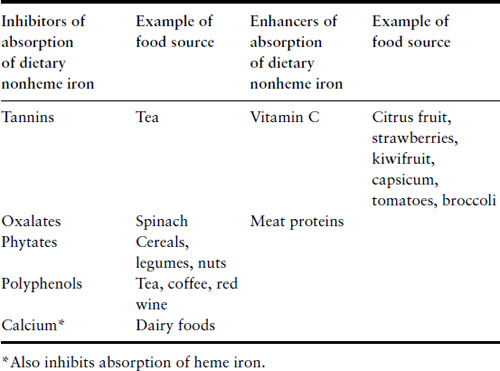
Iron deficiency itself leads to increased absorption of dietary iron and there is also increased absorption during pregnancy. Even with these physiologic changes, women are unable to maintain a positive iron balance during pregnancy and it is not surprising that at least 40% of women will be iron deficient by the third trimester. A diet with high iron bio-availability provides an estimated 3–4 mg of iron each day and absorption can increase to 5 mg/day in the third trimester. The most bio-available food source of iron is heme iron that is derived from hemoglobin-containing animal foods. Around 20–30% of heme iron in food can be absorbed compared to absorption of 2–10% of nonheme dietary iron, i.e. from plant foods. Consumption of food containing heme iron will augment the absorption of iron from nonheme sources. Certain foods inhibit absorption of dietary nonheme iron but with the exception of calcium, foods do not affect absorption of heme iron (Table 2.1). Simple dietary advice can help optimize women’s dietary iron intake and reduce their requirements for iron supplementation. However, given the considerable demands for iron in the second half of pregnancy, one can appreciate how depletion of iron stores occurs during pregnancy and understand why many women will require dietary iron supplements. Women who enter pregnancy with iron deficiency and those with a diet with low bio-availability are at much greater risk of developing iron deficiency and anemia.
Table 2.1 Food factors that modify absorption of dietary iron
Dietary iron supplements: targeted or universal?
Studies have consistently shown that anemia and iron deficiency anemia are risk factors for preterm delivery and for small-for-gestational age infants [3]. The adverse impact of anemia is mainly restricted to anemia present in early pregnancy. It is interesting that correction of iron deficiency anemia with iron supplementation has not lead to reductions in these adverse fetal outcomes; however, the majority of clinical trials were carried out in women who were not anemic early in pregnancy thereby excluding those women who were likely to have shown most benefit from intervention [4]. Similarly, routine iron supplementation in pregnant women who are not iron deficient has not been shown to impact on fetal outcome although it does improve maternal iron stores and hemoglobin levels [5]. Given the absence of evidence of benefit of routine iron supplementation in pregnancy and the potential for adverse effects such as increasing oxidative stress, iron supplementation should be directed to those women who have iron deficiency or IDA.
Despite lack of evidence of improved pregnancy outcome, the US Centers for Disease Control and Prevention (CDC) recommend 30 mg/day elemental iron supplements (e.g. 150 mg of ferrous sulfate) for all pregnant women, with higher doses in women who are iron deficient or have IDA. Although treatment of iron deficiency and iron deficiency anemia does not impact on fetal outcome, correction of maternal anemia and iron deficiency contributes to improved maternal well-being and mortality outcomes. Women who are anemic should be treated to restore the hemoglobin to normal levels in preparation for delivery. Hemorrhage complicating delivery is likely to pose more of a problem for a woman who has pre-existing anemia. In addition, correction of anemia will improve maternal well-being. Iron deficiency even in the absence of anemia can cause maternal symptoms such as fatigue and restless legs and supplementary iron can alleviate these symptoms. Iron deficiency may also have detrimental effects on the immune system and reduce healing.
Commonly available iron supplements are usually the ferrous form of iron coupled with inorganic salts – ferrous fumarate, ferrous sulfate and ferrous gluconate, containing 33%, 20% and 11% elemental iron, respectively; 300 mg of each of these formulations therefore contain approximately 100 mg, 60 mg and 33 mg of elemental iron respectively. Ferrous sulfate is the most readily absorbed form but causes more gastric irritation than the other forms. A typical dose for treatment of IDA would be ferrous sulfate 325 mg PO two to three times a day taken, if possible, on an empty stomach. There is little capacity for the gastrointestinal system to absorb doses significantly above this level. Vitamin C supplementation or consuming the iron supplementation with red meat will increase absorption of supplemental iron. Simultaneous intake of calcium supplementation will decrease iron absorption. Side effects of iron supplements include nausea, vomiting, constipation, and dark stools and are dose related. Hemoglobin levels rise within 2– 3 weeks of starting iron therapy and one of the most common causes of lack of response is noncompliance, mainly because of side effects. In such cases it may be helpful to prescribe a formulation that has a lower concentration of elemental iron to reduce side effects and improve compliance.
Of note, high hemoglobin later in pregnancy is also associated with adverse pregnancy outcome, which is likely to reflect the plasma volume contraction that occurs in placental insufficiency syndromes such as pre-eclampsia [6].
Diagnosis of iron deficiency and iron deficiency anemia
While the functional compartments of iron are contained in hemoglobin and myoglobin, ferritin is the predominant storage compartment for excess iron. Transferrin is the transport molecule that shuttles iron between the storage and functional compartments. Serum ferritin generally provides the most easily assessable measure of total body iron stores. However, ferritin is an acute-phase protein that will increase in the presence of acute or chronic inflammation or infection so will be unreliable in these clinical circumstances. A reduction in serum ferritin is usually the earliest indication of iron deficiency. Low iron stores will reduce the amount (saturation) of iron bound to transferrin, the iron transport protein, so that its capacity to bind iron is increased. A low transferrin saturation or increased total iron-binding capacity is often present in iron deficiency but on their own are unhelpful in diagnosis of iron deficiency because of rapid fluctuations in levels. A newer, more accurate laboratory measure of iron deficiency is the soluble form of the cellular transferrin receptor that binds and internalizes transferrin, thus facilitating intracellular transportation of iron. The solubilized serum transferrin receptor is a truncated form of the receptor that increases in concentration when iron deficiency develops, but only once the storage compartment of iron is exhausted and depletion of the functional iron compartment begins. Measurement of serum transferrin receptor is particularly useful in diagnosing iron deficiency in anemia of chronic disease where the ferritin level increases as an acute-phase response. Not all laboratories provide routine analysis of the serum transferrin receptor.
In pregnancy, the serum ferritin is the most useful measure of iron deficiency. Transferrin levels increase during pregnancy, making measurement of the total iron-binding capacity unhelpful in the diagnosis of iron deficiency.
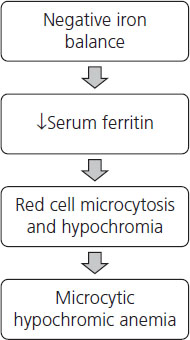
Continued iron deficiency leads to depletion of the functional compartment of iron that initially leads to production of microcytic, hypochromic red cells and ultimately development of anemia, i.e. low hemoglobin and hematocrit (Figure 2.1).
Assessment of iron status in pregnancy
Assessment of iron status and identification of women at risk of developing iron deficiency in pregnancy are an essential part of antenatal care. Booking bloods traditionally include complete blood count (CBC) that can identify anemia and microcytic (cells with a low mean cellular volume or MCV), hypochromic red blood cells in women who already have a significant negative iron balance. These women require urgent treatment to restore iron stores and reverse anemia and should receive relatively high-dose inorganic iron supplementation. Intravenous iron supplementation with iron polymaltose should be reserved for those women who are unable to absorb iron due to gastrointestinal disease or surgical resection. In addition, parenteral iron may be considered for the unusual case where a woman with severe iron deficiency anemia is seen for the first time late in pregnancy and is unlikely to have sufficient time for oral iron replacement to be effective. Parenteral iron is available as iron dextran, iron sucrose, ferric gluconate complex and iron polymaltose. Severe adverse drug reactions including death from anaphylaxis are reported with iron dextran but appear to be far less common with the other forms of parenteral iron. Iron can also be given by intramuscular injection; however, not only is absorption variable but this method of administration leaves permanent stains on the skin. Blood transfusion should be reserved for women with severe symptomatic anemia or other medical conditions such as cardiac disease that will be compounded by maternal anemia.
Measurement of serum ferritin with booking bloods is not a universal recommendation but does provide the opportunity to identify women with marginal iron stores who are likely to become iron deficient earlier in pregnancy. Iron deficiency is diagnosed with a serum ferritin of <12–15 µg/L (12–15 ng/mL or 27–33 pmol/L) in the absence of inflammation.
Vitamin B12: cobalamin
Vitamin B12 belongs to the cobalamin family of compounds which together with folate are a vital part of the so-called one-carbon metabolism cycle that provides methyl groups required for synthesis of DNA, formation of myelin and neurotransmitters such as dopamine. Strictly speaking, vitamin B12 refers only to cyanocobalamin, a form of cobalamin that exists only in vitro. For clarity, the term cobalamin will be used in this text to refer to all members of this vitamin group including vitamin B12.
Cobalamin (Cbl) in humans is derived entirely from the diet, from vegetable sources via cobalamin-producing bacteria on leguminous plants and from animal sources through the cobalamin present in muscle and tissues. The major dietary source of Cbl is animal protein with fish, animal muscle, milk products and egg yolk providing about 1–10 µg cobalamin/100 g wet weight of food. Cobalamin is very effectively stored with total body stores of 2–5mg in adults. It takes about 3–4 years to deplete cobalamin stores if dietary intake is interrupted but the deficiency state takes longer to manifest due to enterohepatic circulation of cobalamin.
Cobalamin in food undergoes peptic digestion at acid pH in the stomach and is released when it initially binds to R-protein (haptocorrin) prior to its passage to the duodenum. There, pancreatic proteases degrade the R-proteins, allowing transfer of Cbl to intrinsic factor (IF) produced by gastric parietal cells. Cbl bound to IF then continues down the small intestine to be bound by specific IF receptors on the terminal ileum to deliver to plasma. Cbl in plasma is wholly bound to transport proteins, transcobalamins, with 10–30% bound to transcobalamin II and most of the remainder to transcobalamin I. Lysosomal transfer of Cbl to the intracellular compartment provides Cbl that is modified and acts as a co-enzyme for the intracellular enzymes methylmalonyl-CoA mutase and methionine synthetase required for one-carbon metabolism [7].
Cobalamin deficiency
Cobalamin deficiency can occur due to inadequate dietary intake, defective absorption or, rarely, defective plasma transportation. Given the availability of cobalamin in food sources of animal origin, it is clear that dietary deficiency will mainly be a problem for individuals who adhere to a strict vegan diet, i.e. one that contains no animal products. However, low levels of B12 are reported in lacto-vegetarians (diet includes dairy products) and also ovo-lacto-vegetarians (diet includes dairy products and eggs) [8]. The most common cause of defective absorption is likely to be gastric achlorhydria that results in the loss of the acidic gastric milieu essential for initial release of Cbl from food sources and reduced transfer of Cbl to binding proteins in the stomach. Achlorhydria can be the result of drug treatment with gastric acid pump inhibitors such as omeprazole but also occurs with aging. Pernicious anemia is an autoimmune disorder in which the autoantibodies are formed to the gastric parietal cells that produce intrinsic factor, thus preventing transportation and transfer of Cbl to the terminal ileum for cellular uptake. Other causes of cobalamin deficiency include gastric or intestinal surgery, blind loop syndrome.
Cobalamin requirements and cobalamin deficiency in pregnancy
A mixed Western diet provides about 5–7 µg/day of Cbl, well above the recommended daily intake of 2 µg/day for adults that increases to 2.2–2.6 µg/day during pregnancy and lactation. Given the minimal fetal Cbl requirements of 60– 100 µg during pregnancy and a relative abundance of dietary cobalamin and maternal stores of around 3000 µg, it seems unlikely that cellular Cbl deficiency will develop in pregnant women who eat a mixed diet so that supplementation of dietary vitamin B12 is unlikely to be necessary. However, a lower dietary cobalamin intake in women who eat ovo-lacto, lacto-vegetarian or vegan diets will result in lower cobalamin stores and dietary intake may not meet the recommended daily requirement [9].
These factors and therefore the potential for cellular deficiency of cobalamin and its resultant effect on DNA synthesis are important when interpreting serum vitamin B12 results during pregnancy. A number of studies have reported a reduction in serum B12 levels during pregnancy but whether this represents a true cellular deficiency of B12 is not clear. The low levels may represent no more than the dilutional effect of plasma volume expansion on serum B12 levels. In contrast to the assessment of iron stores, there is no ferritin equivalent to confirm that there is insufficient cobalamin available for essential cellular functions. However, increases in other intermediates in the one-carbon pathway that require cobolamin and folate have been shown to support a diagnosis of deficiency of one of these vitamins. Typically, cobalamin deficiency will lead to increased levels of methylmalonic acid (MMA) and homocysteine whereas folate deficiency will lead to increases in homocysteine levels. However, the pregnancy-related reduction in homocysteine levels makes interpretation of laboratory measurements of these metabolites difficult during pregnancy. The hematologic effects of Cbl deficiency initially include development of red cell macrocytosis (an elevation in mean cell volume (MCV)) and eventually megaloblastic anemia with characteristic blood film features (Box 2.1). It is important to note that blood film features cannot distinguish between the megaloblastic anemias caused by cobalamin or folate deficiency. Neurologic sequelae include sensory disturbances and confusion.
Box 2.1 Hematologic features of B12 and folate deficiency
- Oval macrocytes
- Hypersegmented neutrophils
- Howell-Jolly bodies
- Thrombocytopenia
- Red cell fragments and tear drop red cells
In clinical practice, screening measurement of serum B12 levels should be restricted to women with significant gastric resections or a low dietary intake of cobalamin, i.e. women on ovo-lacto, lacto-vegetarian or vegan diets. Otherwise testing should be limited to patients with unexplained elevations in MCV on a complete blood count. If reduced serum B12 levels are detected in vegetarian women, they should be offered oral B12 supplements. As their low levels are due to a low dietary intake and not malabsorption, parenteral cobalamin is not essential. However, some women may prefer a single intramuscular injection of 1000 µg of hydroxycobalamin that may be sufficient for the entire pregnancy. Patients with B12 deficiency due to pernicioius anemia or gastrectomy may be treated with 1000 µg every day for 1 week, followed by 1000 µg every week for 4 weeks and then 1000 µg every month thereafter.
Breastfeeding
An adequate dietary intake of cobalamin is essential during breastfeeding to ensure sufficient quantities of cobalamin for the developing infant. Megaloblastic anemia, neurologic abnormalities and developmental delay have been described in breastfed infants of strictly vegetarian mothers and dietary supplementation is essential [10]. Malabsorption of cobalamin can also occur in patients after gastric bypass surgery and these women may require parenteral supplementation of B12 to prevent B12 deficiency in their breastfed infants [11].
Folate
Folate is the generic term for the vitamin that is an essential co-enzyme in the one-carbon metabolism cycle that provides methyl groups required for DNA synthesis and cell division. Synthetic folic acid, used in vitamin preparations and for food fortification, is a monoglutamic acid and is the most active form of the vitamin. It is also the most readily absorbed and has 100% absorption and bio-availability when taken on an empty stomach that is reduced to about 85% when taken with food. Food sources of folate provide the polyglutamate form of the vitamin that is much less readily absorbed, with ≤50% bio-availability. Food sources of folate include green leafy vegetables, mushrooms, yeast and fruit, with high levels present in animal protein such as liver and kidney. Ingested folate is absorbed via specific transport mechanisms in the small intestine and in addition to dietary folate, normal serum folate levels are maintained by the enterohepatic circulation. Cellular transport of folate is mediated by specific folate receptors. Folate is stored in the liver with total body stores of only 8–20 mg, which is small relative to recommended daily requirements, i.e. 100 µg/day (nonpregnant). Serum folate levels fall within a few days of interruption of dietary folate intake even though folate stores are not exhausted. Folate is incorporated into red blood cells early in their production and given their ≈120 day lifespan, red blood cell folate levels will give an indication of folate intake for the 2–3 months prior to blood sampling and red cell folate levels are thought to more accurately reflect total body folate stores than serum levels.
Folate requirements and supplementation during pregnancy
Daily folate requirements increase during pregnancy to 600 µg/day and 300 µg/day during lactation, the additional folate being required for nucleotide synthesis, cell division and increased red cell production. Given the relative small size of folate stores, ongoing severe reduction in dietary folate intake or increased utilization of folate will manifest within a few months [7].
The efficacy of preconceptual folic acid supplementation for primary and secondary prevention of neural tube defects (NTD) is well established. Daily supplements of 0.4 mg folic acid are recommended for low-risk women and 5 mg daily for women at higher risk. Despite this, a significant reduction in the incidence of NTD was not reported until mandatory folate fortification of foods was introduced in 1998. Closure of the neural tube occurs early in gestation, at around 21–28 days post conception, perhaps only 1–2 weeks after an expected missed period. For the many women for whom pregnancy is an unplanned event, this important opportunity for preconceptual folate supplementation would be missed [12].
In addition to folic acid supplementation for prevention of NTD, women with a low dietary intake of folate may require ongoing supplementation to prevent development of anemia. As with Cbl deficiency, the initial hematologic manifestation of folate deficiency is usually development of macrocytosis and if left untreated, megaloblastic anemia ensues.
Folate deficiency: laboratory investigation
Investigation of folate deficiency includes CBC, measurement of serum and occasionally red cell folate levels. The serum folate level is less expensive than the red cell folate level and reflects recent dietary intake and in an otherwise well person, deficiency is unlikely if serum folate levels are normal. Low serum folate levels are commonly found in hospitalized patients and confirmation of low serum levels by measurement of the more expensive red cell folate is recommended before making a diagnosis of deficiency but otherwise the serum folate alone can be used for diagnosis [13]. Of note, serum folate levels may be increased with cobalamin deficiency as a result of the block in the one-carbon metabolism pathway. Development of macrocytic red cells induced by deficiencies of folate and/or cobalamin may not occur in the presence of co-existing iron deficiency or the presence of a thalassemia or hemoglobinopathy. Careful examination of the peripheral blood smear may reveal other abnormalities suggestive of a deficiency such as hypersegmented neutrophils.
Confirmation of folate deficiency and differentiating it from cobalamin deficiency can be problematic as none of the commonly used laboratory tests is 100% sensitive and specific for diagnosis of a cellular deficiency of either vitamin. Measurement of other intermediates in the one-carbon cycle such as MMA and homocyteine (Hcy) have been suggested to be more specific and sensitive methods of determining a deficiency state and of differentiating between cobalamin and folate deficiency. Blocks in the one-carbon cycle due to deficiency of cobalamin will lead to increased levels of both MMA and Hcy whereas folate deficiency will only increase levels of Hcy. A reduction in Hcy levels in pregnancy due to increased glomerular filtration rates and lack of a defined normal range for pregnancy may limit the utility of these laboratory analytes in pregnancy, apart from the expense and limited availability of the tests themselves [14].
It is worth mentioning here that although macrocytosis may be caused by either or both folate and B12 deficiency, other causes include alcoholism, hypothyroidism, liver disease, hyperlipidemia and some medications (including phenytoin, azathioprine and zidovudine). The macrocytosis that occurs in some individuals who take anticonvulsant drugs is mediated by undefined mechanisms that are not due to folate or B12 deficiency and therefore does not respond to vitamin supplementation.
Summary
Anemia is the most prevalent hematologic abnormality in pregnancy, one that most commonly is caused by factors associated with poverty and social deprivation. It is associated with significant fetal and maternal morbidity and mortality, it can be easily diagnosed and responds quickly to intervention and treatment that is both inexpensive and readily available. Efforts to improve maternal and infant mortality could be addressed by diagnosis and treatment of anemia that on a global scale will have most impact in resource-poor countries but that could also lead to significant improvements in pregnancy outcome and maternal well-being for less advantaged women in more industrialized countries.
The hemoglobinopathies and thalassemias
Hemoglobinopathies and thalassemias are structural and quantitative abnormalities of hemoglobin. Together, they are the most common single-gene disorders affecting humans, found in almost 5% of the world’s population. Originally described in people from the Mediterranean and South East Asia, it was later recognized that hemoglobinopathies were common in people from Africa, the Indian subcontinent and the Pacific, with marked differences in prevalence existing within regions. The original geographical distribution of hemoglobin variants is thought to be due to the protective effect that the heterozygous state confers against malaria, the mosquito-borne disease that is a major cause of mortality and morbidity in these regions. Demographic changes have resulted in major increases in the number of individuals with hemoglobinopathies and thalassemia in North America and Northern Europe [15] Such changes require that, globally, clinicians involved in obstetric care develop an understanding of the potential health consequences of these disorders for mother and child.
Structure of hemoglobin
Hemoglobin is the iron-containing compound in red blood cells that is responsible for oxygen transfer in vertebrates and mammals, including humans. The hemoglobin molecule is a tetramer composed of two different pairs of globular protein subunits, i.e. globins, each of which is covalently attached to a nonprotein heme moiety that carries iron and is responsible for binding oxygen. In adult life the predominant hemoglobin type is Hb A, made up of two α and two β chains (α2β2), accounting for around 95–97% of adult hemoglobin, with 1.5–3.5% Hb A2 (α2γ2) and HbF (α2δ2) making up the remainder. Different globin chains are expressed in embryonic and fetal development so that in embryogenesis the hemoglobins are Hb Gower 1 (ζ2ε2), Hb Gower 2 (α2ε2) and Hb Portland 1 (ζ2δ2). By 8–9 weeks gestation, the embryonic ζ – and ε-globin chains are no longer detectable and Hb F (α2δ2) is the predominant hemoglobin during fetal development. Synthesis of the β-globin chain begins from around 7 weeks gestation with Hb A accounting for around 10% of total hemoglobin at birth, reaching normal adult levels by 6 months to 1 year of age. Chromosome 16 is the site of the α-globin gene cluster composed of two α-globin genes and a single ζ-globin gene with the non-α chains, β, ε, and γ encoded by single genes on chromosome 11. Normal individuals therefore express four α-globin genes and two β-like genes. Deletions or mutations of the α- or β-globin genes will lead to α- or β-thalassemia, respectively, with most hemoglobinopathies occurring as a result of mutations affecting the β-globin gene cluster [16].
Thalassemias and hemoglobinopathies can be categorized in a number of ways. Firstly, according to the severity of clinical manifestations, i.e. the phenotypic expression, ranging from the most severe (thalassemia major) to that of moderate severity (thalassemia intermedia) to a mild or clinically silent phenotype (thalassemia minor). Thalassemias can be also grouped by whether the mutation affects the α- or β-globin chain with the specific mutations or deletions described.
Disorders of α-globin synthesis
α-Thalassemia results from reduced production of α-globin chains due mainly to deletions of one or more of the four α- globin genes (Figure 2.2). The length of the deletion determines whether one or both α-globin genes are missing from the same chromosome. The South East Asian (SEA), Philippino (FIL), Thai (THAI) Mediterranean (MED) and 2 (α)20.5 genotypes cause loss of both α-chains from a single chromosome, leading to cis 2-gene deletion α-thalassemia known as α0-thalassemia (cis refers to the fact that the two mutations are both on the same chromosome). These mutations are particularly common in South East Asia, the Mediterranean and the Middle East. Single α-globin gene deletions are also prevalent in these geographical areas but also in the Pacific region and include 2 α4.2 and 2 α3.7 and lead to one-gene deletion denoted by α+- thalassemia. Nondeletional α-thalassemia also occurs, the most common defect being Hb Constant Spring where the mutation leads to formation of unstable extended α-globin chains. Carrier rates of α-thalassemia show marked geographical variation, with rates of up to 25% reported in Thailand, around 3% in China, with the α+-thalassemia described in 10–15% of people of Pacific origin [17].
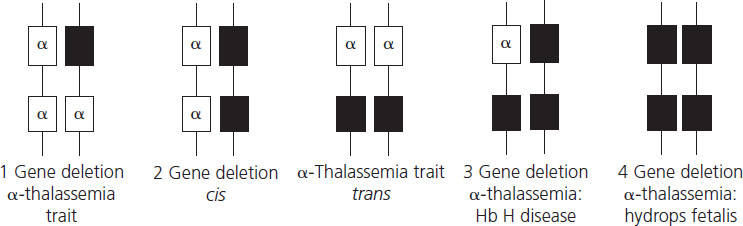
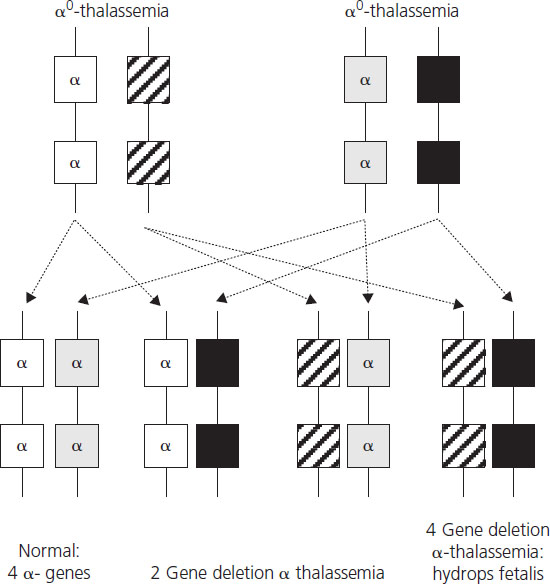
Hydrops fetalis, the most severe form of α-thalassemia, results from deletion of all four α-globin genes. Although the literature reports a small number of survivors with this condition [18], it is usually lethal in utero as α-globin chains are required for formation of fetal hemoglobin from very early in fetal life. Absence of α-globin results in the formation of Hb Barts made by tetramers of the γ-globin chain (γ4), the fetal equivalent of the β-globin chain. Hb Barts has a very high oxygen affinity and does not release oxygen to fetal tissues, leading to severe hypoxia and ultimately fetal hydrops. Hydrops fetalis most commonly results from inheritance of α0-thalassemia from each parent (Figure 2.3). The geographical distribution of these genotypes means that hydrops fetalis is therefore particularly common in South East Asia where it poses a major health problem.
Figure 2.3 Potential inheritance patterns in offspring of parents with α0-thalassemia traits demonstrating a one in four chance of development of hydrops fetalis due to absence of four α-chains.
Hb H disease is due to loss of three α-globin genes (2 2/2 α) (see Figure 2.3). Deficiency of α-globin chains with excess of β-globin chains leads to formation of Hb H, a tetramer of β-globin (β4) which is relatively unstable and forms intracellular precipitates that ultimately shorten red cell survival. Hb H disease generally exhibits a mild clinical phenotype with mild anemia that is not usually transfusion dependent although in some patients anemia is more severe, requiring transfusion.
Two-gene deletion α-thalassemia or α-thalassemia trait can be due to deletions of α-globin genes from the same chromosome (2 2 /α α) or to single gene deletions from both chromosomes (2 α/2 α). α-Thalassemia trait is phenotypically mild with microcytic, hypochromic red cell indices and occasionally mild anemia. It is not possible to distinguish between the two types of two-gene deletion α-thalassemia using basic hematologic indices and special molecular studies are required.
Single gene deletion α-thalassemia or α+-thalassemia (2 α/α α) is clinically silent and may present only with mild red cell microcytosis and hypochromia. It is the predominant form of α-thalassemia trait in people of Pacific origin but is in Africa, the Mediterranean and South East Asia.
Disorders of β-globin synthesis
Disordered synthesis of the β-globin chain usually results from point mutations of the β-globin gene, of which over 200 different mutations have been described. β-Thalassemia is caused by mutations that lead to reduced (β+) or absent (β0) production of structurally normal β-globin chains. In contrast, the hemoglobinopathies result from mutations that produce usually normal amounts of structurally abnormal or variant hemoglobins.
β-Thalassemia
β-Thalassemia trait, prevalent in the Mediterranean, North Africa, the Middle East, the Indian subcontinent and South East Asia, is due to inheritance of one of nearly 200 single β-globin gene mutations, usually a point mutation or single amino acid change that leads to reduced production of a normal β-globin chain. It exhibits a clinically mild phenotype but with microcytic, hypochromic red cells seen in the peripheral blood smear. Inheritance of two abnormal β-globin genes leads to thalassemia major or thalassemia intermedia, depending on the combination of β-globin gene mutations inherited. One of the most severe clinical phenotypes occurs in individuals homozygous for β0 mutations resulting in complete absence of β-globin chain production. This presents in the first year of life with severe anemia, hepatosplenomegaly, bone deformities and growth delay. Individuals who are compound heterozygotes for β0 and β+ mutations show variable clinical manifestations ranging from those with severe transfusion-dependent anemia to those who require only periodic transfusions. More recently, it has been recognized that β-thalassemia major or intermedia is associated with an increased risk of venous thromboembolism, especially in individuals who have had a splenectomy. The anemia in β-thalassemia is due to ineffective erythropoiesis and hemolysis, both of which are the result of an excess of α-chains. There is considerable variation in the prevalence of β-thalassemia among different countries, ranging from 2–3% in the Middle East to 17% in Cyprus. Also marked variations are described within individual countries; for example, in Greece the prevalence ranges from 6% to 19% and in Italy from 7% to 19%. Clinically significant interactions of β-thalassemia trait with other hemoglobin variants, such as Hb S and Hb E, are described under the individual hemoglobinopathies.
Hemoglobinopathies
Of the greater than 200 different β-globin gene mutations, only a very small number, i.e. Hb E, Hb C, Hb S, and Hb Lepore, cause clinically significant abnormalities. This occurs most commonly in the compound heterozygous state with other β-chain abnormalities but importantly sickle cell disease develops in individuals homozygous for Hb S. These mutations show quite marked geographical variability: Hb E is particularly prevalent in South East Asia, with carrier rates of 20–60%, and Hb S and Hb C are common in Africa with carrier rates of 12% and 3%, respectively. In the heterozygous or carrier state these hemoglobinopathies do not cause clinical abnormalities but a high carrier frequency in certain populations leads to perpetuation of severe thalassemia syndromes in the children of asymptomatic carriers. Carriers are frequently undiagnosed when embarking on pregnancy, do not receive pre-pregnancy or antenatal counseling or may choose not to have antenatal testing for social or cultural reasons.
Hb E
Hb E is caused by a single point mutation at position 26 on the β-chain (βE) where glutamine is replaced by lysine, resulting in reduced synthesis of the β-globin chain. Individuals who are heterozygous or even homozygous for HbE are completely asymptomatic and its clinical relevance stems from the consequences that occur when this very common hemoglobin variant combines with β-thalassemia trait. Compound heterozygotes for HbE and β-thalassemia have a wide range of clinical manifestations from mild anemia to the most severe form of thalassemia major. Such marked phenotypic variability is not well understood and, in particular, does not seem to be related to the specific mutation responsible for the β-thalassemia trait. So even among individuals with the same β-chain mutation in combination with Hb E, there are major differences in the clinical severity of the thalassemia that results.
Hb C
Hb C is caused by a single point mutation at position 6 on the β-chain where glutamine is replaced by lysine, resulting in a variant Hb (βc) that is less soluble than Hb A and also has a lower oxygen affinity. Thought to have originated in West Africa, the heterozygous state has an overall prevalence of around 3% that reaches as high as 40% in Burkina Faso and 50% in Ivory Coast. Individuals heterozygous for Hb C are clinically well and homozygotes exhibit mild, chronic hemolytic anemia. Combinations of Hb C with β-thalassemia or other variant hemoglobins, especially Hb S, lead to clinically significant disorders. Compound heterozygotes for Hb C and β0-thalassemia have moderately severe anemia with splenomegaly whereas those with Hb C and mild β+-thalassemia have mild to moderate hemolytic anemia. The clinical implications for individuals who are compound heterozygotes for Hb C and Hb S are discussed later.
Hb Lepore
Hb Lepore is a hemoglobin variant that results from creation of a fusion δβ-globin gene, due to abnormal cross-over during meiosis. There is inefficient synthesis of the δβ-chain of Hb Lepore that leads to a thalassemia phenotype. Individuals heterozygous for Hb Lepore are asymptomatic with microcytic, hypochromic red blood cell indices similar to those found in β-thalassemia trait. The homozygous state varies between transfusion-dependent thalassemia major and thalassemia intermedia. Compound heterozygotes for Hb Lepore and β- thalassemia manifest as thalassemia major.
Hb S
Hb S or sickle cell trait results from replacement of glutamine by valine on position 6 of the β-globin chain (βS). It is particularly common in sub-Saharan Africa but also occurs in the Indian subcontinent, the Middle East and the Mediterranean. The carrier frequency in African Americans is 8%. The deleterious effects of Hb S result from polymerization of Hb S that occurs when the Hb molecule is deoxygenated, leading to reduced deformability of the red cell and increased adhesion to endothelial cells. This results in reduced red cell survival due to hemolysis and also vaso-occlusive events, especially in the low oxygen environment of the capillary circulation. The polymerization of Hb S is particularly slowed by the presence of Hb F but also to a lesser degree by Hb A2 and Hb A [19].
Sickle cell disease
Red cell sickling does not occur in individuals who have sickle cell trait except in conditions of extremely low oxygen tension, such as travel by air, especially in unpressurized cabins, vigorous exercise or high fever. Patients homozygous for Hb S (α2βS2) have sickle cell disease (SCD) although, as seen later, SCD also occurs in patients who are compound heterozygotes for Hb S and other β-chain abnormalities such as β-thalassemia trait. The major clinical manifestations of SCD are due to vaso-occlusive events that either present acutely as a sickle cell or vaso-occlusive crisis or as the result of chronic vascular occlusion. Most individuals have chronic hemolysis with a hemoglobin in the order of 6–9 g/dL but symptomatic anemia is uncommon as patients compensate well for their degree of anemia, especially as Hb S has a relatively low oxygen affinity. The skeletal and growth abnormalities seen with β-thalassemia major do not develop and overall patients go through normal puberty. The most significant clinical problems are the result of acute and sometimes life-threatening vaso-occlusive sickling crises that can affect bones, lungs, the brain, spleen, liver and in men can cause recurrent priapism. Crises can be provoked by cold, dehydration and infection but are self-limiting, usually lasting a few days. About 20% of patients with SCD suffer from recurrent severe events and 50% have about one crisis per year. Vaso-occlusion of the bone marrow leads to severe bone pain usually requiring narcotic analgesia. Lung crises or acute chest syndrome, a common cause of mortality, presents with hypoxia, dyspnea and pulmonary infiltrates on X-ray due to sequestration of sickled cells in the pulmonary circulation. Brain syndrome with stroke or hemorrhage is described in 11% of patients by the age of 20 years and in 24% by the age of 45 years. Manifestations of chronic vascular occlusion include aseptic necrosis of the femoral or humeral head, pulmonary hypertension, leg ulcers, proliferative retinopathy and progressive renal failure. Sepsis is an important cause of morbidity and mortality for individuals with SCD at all ages, in particular due to Strep. pneumoniae, Salmonella and Klebsiella, and pneumococcal vaccination is recommended.
The pathophysiology of vaso-occlusion in SCD is more complex than simple mechanical obstruction by rigid, sickled red cells. Sickled red cells are also more likely to adhere to vascular endothelium due to their innate properties and also to increased expression of inflammatory markers and adhesion molecules on leukocytes and the endothelial surface. Ischemia-reperfusion injury is thought to be a major contributor to the proinflammatory state in SCD [20].
Hb F inhibits polymer formation of the βS-globin chains so that individuals with increased levels of Hb F have a less severe clinical phenotype of SCD. The level of Hb F has been shown to be a prognostic factor for complications of SCD. Certain ethnic subgroups, for example people from India and Saudi Arabia, record higher levels of Hb F and a very mild clinical phenotype. As mentioned previously, SCD also occurs when Hb S is co-inherited with other abnormalities of the β-chain. Compound heterozygotes of β-thalassemia and Hb S show a very variable clinical phenotype ranging from mild disease in patients of African origin with Hb S-β+thalassemia to more severe forms of when combined with β0-thalassemia and even β+-thalassemia from other ethnic groups. In general, it is not possible to predict the clinical course of SCD in compound heterozygotes of β-thalassemia and Hb S on the basis of the β-thalassemia genotype, thus making genetic counseling particularly problematic. Individuals who inherit both Hb S and Hb C have milder, chronic hemolytic anemia and although they experience fewer sickle cell crises, they are more prone to vaso-occlusive osteonecrosis and also retinal disease.
Treatment of SCD
Increased levels of Hb F are associated with a less severe clinical phenotype and studies have shown that treatment with the cytotoxic agent hydroxyurea increases expression of Hb F, leading to a reduction in frequency of sickling crises, acute chest syndrome and overall mortality [21]. Treatment of an acute crisis involves maternal oxygen supplementation, rapid relief of pain (usually requiring opioid analgesics) and maintenance of good oral fluid intake with intravenous fluids if required. Blood transfusion is not routinely required in an uncomplicated sickling crisis but in severe chest crises, cerebrovascular events or multiorgan failure, exchange transfusion is indicated [22].
Interaction of α-thalassemia with β-chain abnormalities
The clinical consequences of the thalassemias result from an imbalance of α-chains relative to β-chains. Given the geographical distribution of these mutations, co-inheritance of both α-thalassemia traits and β-thalassemia major or minor is therefore not surprising. When this occurs, a less severe clinical phenotype is present as there is an overall reduction in α-chains relative to β-chains which protects against the effects of excess globin chains. When α-thalassemia is inherited with β-chain hemoglobin variants, lower amounts of the abnormal Hb are formed overall as the α-chains preferentially bind to normal β-chains.
Diagnosis of thalassemias and hemoglobinopathies
The diagnosis of thalassemias and hemoglobinopathies requires a combination of laboratory tests. In the absence of iron deficiency, the finding of microcytic, hypochromic red cell indices on an automated CBC are strongly suggestive of the diagnosis of a hemoglobin disorder. Mean cell volume (MCV) describes red cell size (normal range ≈80–100 fL) with microcytosis referring to small red cells. An MCV <78 fL is suggestive of a thalassemia or hemoglobinopathy although the MCV does increase with storage of blood samples and can lead to falsely high results. The mean corpuscular hemoglobin (MCH) is a more stable analytic variable so provides more reliable results. Testing for hemoglobinopathies is recommended if the MCH is less than 27 pg [23]. The peripheral blood smear should be examined for abnormal red cell morphology, such as target cells (red blood cells characterized by a densely stained center surrounded by a pale unstained ring that is encircled by a dark irregular band), stippled cells (small basophilic inclusions distributed throughout the cytoplasm), sickle cells (the characteristic sickle-shaped cell), nucleated red cells and red cell inclusions (round dense bodies of variable size, usually single and staining similarly to a nucleus, also known as Howell Jolly bodies). The Hb H inclusions of α- thalassemia (globular bodies scattered throughout the red cell cytoplasm) can be generated in vitro by using an oxidative dye such as new methylene blue and the peripheral blood smear reviewed to determine their presence and the number of red cells affected. Hemoglobin electrophoresis is routinely used to detect the presence and quantity of normal and abnormal hemoglobins although high-performance liquid chromatography (HPLC) is a useful tool for this purpose. And serendipitous discovery of variant hemoglobins is becoming more frequent as HPLC is also used to quantify glycosylated Hb (Hb A1c) in people with diabetes. Specific laboratory tests are used to detect sickle cell trait and disease. A diagnostic pathway for investigation of hemoglobinopathies and thalassemias is given in Figure 2.4 with a summary of helpful laboratory findings with specific disorders listed in Table 2.2.
Figure 2.4 Diagnostic pathway for thalassemias and hemoglobinopathies. CBC, complete blood count; MCH, mean corpuscular hemoglobin.
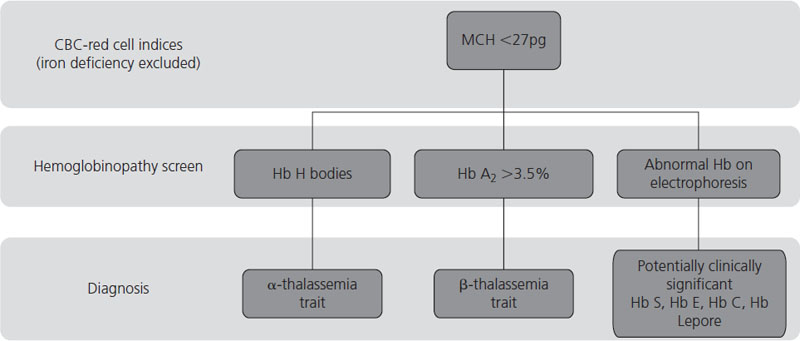
Table 2.2 Hematologic, blood film and hemoglobinopathy findings for certain thalassemias and hemoglobinopathies
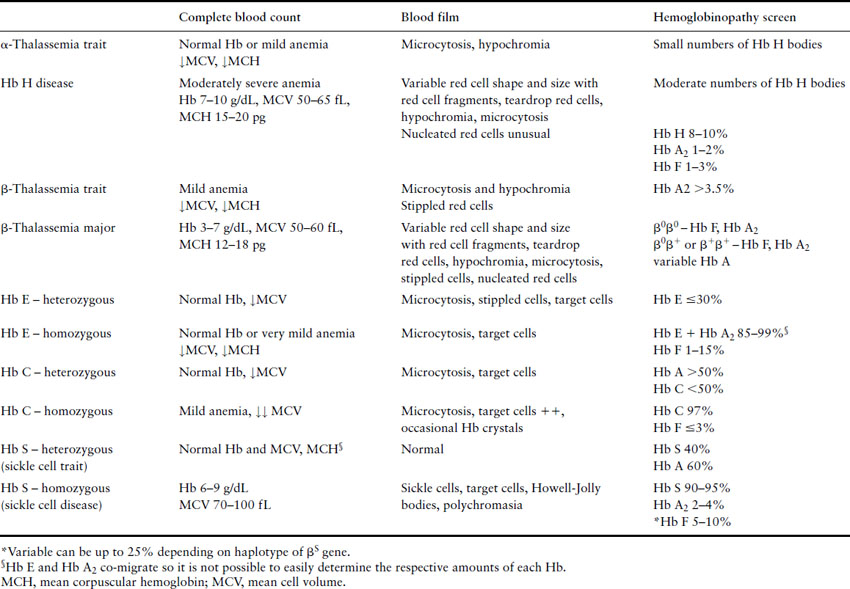
Pregnancy and the thalassemias and hemoglobinopathies
The relevance of the thalassemias and hemoglobinopathies in obstetrics relates to the potential impact on fertility and pregnancy complications in women who have these disorders, especially those who have clinical manifestations. Of equal importance, given the prevalence of these hemoglobin mutations, is the risk of abnormal genes being passed from parents to children resulting in the perpetuation of clinical disease in the next generation. Pre-pregnancy counseling and antenatal diagnosis are key aspects of management in these conditions.
Impact of the thalassemias and hemoglobinopathies on fertility and pregnancy outcome
Thalassemia major
In the developed world, the major clinical complications of transfusion-dependent β-thalassemia result from iron overload due to the disease itself and also secondary to blood transfusions. Chelation therapy (the use of binding agents to remove heavy metals from the body) plays a vital role in minimizing iron overload but if inadequate, leads to iron deposition and ultimately dysfunction of the liver, heart and endocrine organs. Even with tertiary care, the majority of adult patients with transfusion-dependent β-thalassemia have hypogonadotropic hypogonadism that may not always respond to gonadotropins. Other important endocrine disorders relating to pregnancy include diabetes and hypothyroidism as well as liver disease or transfusion-transmitted viral infections such as HIV and hepatitis C.
Tuck described the outcome of 29 pregnancies in 22 women with transfusion-dependent thalassemia major [24]. He suggests that assessment of cardiac function is the most important pre-pregnancy investigation as the presence of cardiac impairment is a poor prognostic factor that is associated with a significant risk of premature death. Both of the maternal deaths in their patient population were due to cardiac failure and they would discourage pregnancy in women with cardiac impairment. Other important pre-pregnancy care includes optimization of diabetic control, thyroid replacement and, importantly, determination of the hemoglobinopathy status of the male partner. Calcium and vitamin D supplements should be given during pregnancy in view of the high incidence of reduced bone mineral density in these patients. Tuck did not report an increased incidence of fetal growth restriction or preterm delivery as has been reported by other centers but put this down to maintaining the maternal hemoglobin above 10 g/dL during pregnancy. He emphasizes the importance of thorough pre-pregnancy assessment to determine maternal risks during pregnancy and whether pregnancy is even advisable, as well as close antenatal monitoring for development of maternal complications.
Sickle cell disease
A large cohort study from birth through young adulthood [25] reported a significantly reduced livebirth rate of 57% (48/94 pregnancies) in 52 women with sickle cell disease compared to a livebirth rate of 89% (128/157) in 68 control women. The difference in outcome was due primarily to an increase in spontaneous miscarriage in women with SCD: 36% (30/94 pregnancies) compared to 10.4% (15/157 pregnancies) in controls. Six stillbirths were reported in the women with SCD compared to only one in the control group. There were no differences in development of pregnancy-induced hypertension, pre-eclampsia or antepartum or postpartum hemorrhage between groups. Other studies have reported high rates of pre-eclampsia in the order of 15–20% in multiparous women with sickle cell disease with histopathologic evidence of placental fibrosis, infarction and thrombosis in the majority of cases. An increased incidence of sickle cell crises is described, particularly in the last trimester of pregnancy and the postpartum period. Women with SCD are advised to ensure they remain well hydrated during pregnancy. The increased risk of infection in SCD should be remembered, especially as it can trigger an acute sickling crisis. Additional folate supplementation (1 mg daily) is required because of chronic hemolysis in SCD. The use of hydroxyurea in pregnancy to increase the levels of hemoglobin F is not justifiable in pregnancy due to the potential teratogenic effects of this agent.
Crises should be managed as in nonpregnant patients but clinicians should be aware that episodes are often more prolonged than those outside pregnancy. Acute crises are managed with aggressive pain control, hydration, oxygen supplementation, continued folate and a careful search for and treatment of any underlying infection. Severe crises may warrant transfusion therapy with the goal being to achieve at least 50% normal hemoglobin, i.e. Hb A.
The role of prophylactic blood transfusions in pregnant women with SCD has been controversial [26]. A randomized trial demonstrated no improvement in maternal and fetal outcome when comparing prophylactic blood transfusions to emergency transfusion for sickle cell crises. Women who received prophylactic red cell transfusions experienced fewer painful crises during pregnancy but women were exposed to four times as many units of blood [27]. In the absence of an improvement in maternal or fetal outcome, prophylactic red cell transfusion during pregnancy is not recommended.
The peridelivery period is associated with a particularly high risk of provoking a sickling crisis. Management of labor includes maintaining adequate hydration and oxygenation and providing good pain relief. Mode of delivery should be determined by obstetric indications.
Hb H disease and thalassemia traits
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


