KEY POINTS
• Anemia is the most common medical complication of pregnancy.
• “Physiologic anemia” of pregnancy is a consequence of disproportionate expansion in red cell mass and blood volume.
• Pregnancy is a state of hypercoagulability. Alteration in hormonal milieu, increase in coagulation factors, and a decrease in anticoagulants and fibrinolysis activity actuate this process.
• Hematologic cancers in pregnancy are rare. Pregnancy does not usually affect the course of the disease, and treatment is generally not withheld during pregnancy.
NORMAL PHYSIOLOGY
Background
• Pregnancy-associated changes (1):
• Increased plasma volume
• Increased formed elements: erythrocyte mass, platelets, and leukocytes
• Adjusted coagulation factors
• Upper and lower limits of these changes in the hematologic elements are somewhat variably defined, but general ranges can be appreciated (Table 19-1) (2).
Table 19-1 Normal Pregnancy Values of the Different Blood Elements
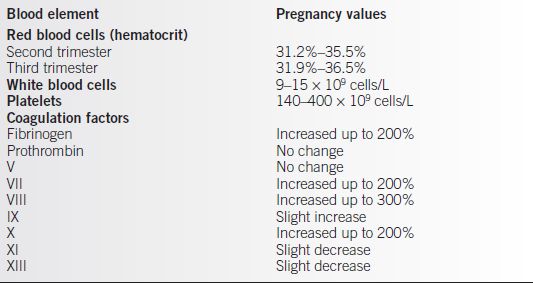
Adapted from Kilpatrick SJ. Anemia and pregnancy. In: Creasy RK, Resnik R, Iams JD, eds. Maternal-fetal medicine principles and practice. 6th ed. Philadelphia: W.B. Saunders, 2009:869–884.
RED BLOOD CELLS
Background
• Increases in both the number of red blood cells (RBCs) and plasma volume occur until near term and then gradually return to prepregnancy levels by 6 weeks postpartum.
• Plasma volume increases to three times that of red cell volume. Expansion begins at 6 weeks and increases significantly until 34 weeks gestation (50% over baseline).
• RBC volume expansion is more gradual than plasma volume from around 10 weeks until term (20% to 30% over baseline).
• A greater increase in plasma volume than red cell volume leads to a decrease in the hemoglobin–hematocrit, resulting in the “physiologic anemia” of pregnancy.
• Multiple-gestation pregnancy is associated with an even larger increase in blood volume.
• An increase in red cell and plasma volume provides for the increased perfusion needs of the fetal–placental unit, accommodates maternal oxygen demands, and gives a margin of safety associated with blood loss during delivery (2).
ANEMIA
Background
• Anemia is the most common medical complication of pregnancy.
• Globally, as many as 41.8% of pregnant women are anemic. The prevalence in the United States has not been well studied; however, WHO approximates 18% in industrialized countries (3,4).
• Rate is influenced by smoking status, high altitudes, race, and chronic medical conditions.
• The most common causes of anemia are nutritional-related iron deficiency and acute blood loss.
• Hgb levels less than 6 g/dL and greater than 14 g/dL are associated with adverse maternal and fetal outcomes (5).
• Recent studies have re-examined the association between maternal anemia and adverse perinatal outcomes. In the setting of moderate to severe anemia, there is an increased risk of prematurity, low birth weight, and fetal death (6–8).
• The inability to expand blood volume or chronic hypoxia may explain the underlying pathophysiology influencing fetal outcomes (6).
• In addition to increased maternal–fetal risk associated with anemia, long-term metabolic and developmental outcomes in children are still being investigated (7).
• Delayed cord clamping following birth has been shown to demonstrate a higher hematocrit in neonates. There is no evidence of maternal or neonatal compromise as once speculated (9).
Definition
• The acceptable lower limit for hemoglobin is 10 g/dL (normal of 14.0 ± 2.0 g/dL).
• Given the normal physiologic hemodilution of pregnancy, the Centers for Disease Control and Prevention defines anemia in pregnancy as follows (10):
• Hgb less than 11.0 g/dL in the first and third trimesters
• Hgb less than 10.5 g/dL in the second trimester
Evaluation
History and Physical
• Signs and symptoms range from subclinical with mild anemia to nonspecific in those with moderate to severe anemia. Symptoms may include:
• Fatigue
• Weakness
• Anorexia
• Headache
• Dyspnea on exertion
• The first prenatal visit should include universal screening for anemia, including questions relating to a history of anemia, personal and family history of bleeding diathesis, and other blood disorders.
Laboratory Tests
• Routine prenatal laboratory studies should include a CBC.
• The hemoglobin/hematocrit should be repeated during the third trimester (about 28 to 32 weeks) and more frequently if indicated.
• Hemoglobin less than 10 g/dL is abnormal in pregnancy and should trigger an evaluation for etiology of anemia (11).
• In the presence of low hemoglobin levels, RBC indices are helpful in deciding the pathophysiology of the anemia.
• Further evaluation may include
• Urinalysis
• Iron studies
• Reticulocyte count
• Peripheral blood smear
• Stool for ova and parasites
• Stool for occult blood (guaiac test)
• Bone marrow studies rarely indicated in pregnancy
• Certain ethnicities should have screening tests for specific conditions.
• African American patients should have a hemoglobin electrophoresis to evaluate for sickle cell trait and thalassemia.
• Patients from the Mediterranean, the Middle East, India, and Southeast Asia are at risk for thalassemia, which also can be identified on hemoglobin electrophoresis.
SPECIFIC ANEMIAS
• Acquired causes of anemia
• Iron deficiency anemia
• Folate deficiency anemia
• B12 deficiency
• Anemia of chronic disease
• Acquired hemolytic anemia
• Aplastic anemia
• Hemorrhagic anemia
Iron Deficiency Anemia
Background
• Most common cause of anemia in pregnancy.
• Antepartum iron deficiency is associated with low iron levels in the infant at 1 year of life but has not been demonstrated to be associated with fetal anemia (10).
Etiology
• Most reproductive-age women have inadequate prenatal iron stores entering pregnancy.
• Factors contributing to iron deficiency include the following:
• Dietary insufficiency, malabsorption, or iron supplement intolerance
• Bleeding prior to or during pregnancy
• Multiple gestation, which increases iron requirements and may be responsible for a greater blood loss at delivery
• Concurrent antacid use may prevent iron absorption
• Poor dietary habits or pica (an appetite for inedible substances, such as clay or dirt)
• Short interpregnancy interval
• Pregnancy exhausts maternal iron stores because of increased maternal blood production and fetal growth needs.
• An approximate iron deficit of 580 mg occurs in pregnancy.
• Conventional diets, even with a focus on iron-rich food, contain about 15 mg of daily iron and do not meet the additional iron requirement of pregnancy.
• Dietary iron intake of 27 mg/day is recommended during pregnancy.
• In total, pregnancy requires approximately 1 g of elemental iron
• 450 mg for RBC expansion
• 270 mg for fetal iron
• 170 mg for basal loss
• 150 mg for delivery loss
• Eighty percent of available functional iron is found in the Hgb of RBCs.
Evaluation
Laboratory Studies
• Iron deficiency anemia (IDA) manifests abnormal lab findings after maternal iron stores are significantly depleted.
• Characterized by a microcytic, hypochromic anemia.
• Decreased mean corpuscular volume (MCV) approximately to a borderline level of 70 to 80 fL (normal = 90 ± 10 fL).
• Serum iron falls below 40 μg/dL.
• Since iron is transported bound to transferrin, the unsaturated total iron-binding capacity (TIBC) rises above 400 μg/dL.
• Serum ferritin (abnormal less than 12 μg/dL) correlates well with bone marrow stores, making a bone marrow examination rarely necessary (12).
 Ferritin is an acute-phase reactant and may be elevated in the presence of inflammation (normal ferritin with normal C-reactive protein is not consistent with IDA) (13).
Ferritin is an acute-phase reactant and may be elevated in the presence of inflammation (normal ferritin with normal C-reactive protein is not consistent with IDA) (13).
 An alternative to ferritin measurement is soluble transferrin receptor (sTfR), which identifies decreased rate of erythropoiesis, levels greater than 4.4 mg/L suggest iron deficiency in tissues (14).
An alternative to ferritin measurement is soluble transferrin receptor (sTfR), which identifies decreased rate of erythropoiesis, levels greater than 4.4 mg/L suggest iron deficiency in tissues (14).
Diagnosis
Differential Diagnosis
• Includes anemia of chronic disease and heterozygous thalassemia
• Thalassemia minor produces a mild anemia with a borderline low MCV but should not exclude the diagnosis of IDA.
• Serum ferritin level, which is highly sensitive and specific for IDA, should help differentiate.
Treatment
Prophylaxis
• In the absence of a laboratory diagnosis of iron deficiency, clinical studies have failed to show a consistent benefit for routine iron supplementation in terms of outcome such as low birth weight or preterm delivery (15–18).
Medications
• Most common oral iron replacement formulations contain inorganic nonheme supplements.
• Limited information is available on organic heme iron supplements but appear to demonstrate better improvement in iron stores postpartum in comparison to placebo or nonheme iron (19).
• Ferrous sulfate 325-mg tablets contain 65 mg of elemental iron.
• Intestinal absorption, approximately 10% of dietary intake, permits absorption only up to 15 mg of iron without signs of iron intolerance.
• Given the limits of daily absorption and the well-recognized gastrointestinal intolerance to iron, TID oral dosing is unnecessary.
• Alternative treatments to oral therapy are parental iron, subcutaneous erythropoietin, and darbepoetin.
• Parental iron formulations are iron dextran (IM/IV), iron sucrose (IV), and sodium ferric gluconate (IV). Iron sucrose and sodium ferric gluconate are better tolerated.
• Parental iron should be considered in setting of oral iron noncompliance or intolerance, malabsorption syndrome, and significant anemia less than 8.5 g/dL.
• No benefit of parental (IV) iron therapy over oral iron therapy when Hgb evaluated 30 days after initiation of therapy (20).
• Reticulocytosis should be observed after 7 to 10 days, and an increase of 1 g/dL should be seen within 14 days after initiation of iron therapy.
• If severe anemia (less than 6 g/dL) is present in the antepartum period, blood transfusion should be considered for fetal and maternal indications.
• Iron supplementation should continue for 6 months after anemia resolves to replace iron stores.
Side Effects
• The dose of elemental iron correlates with the symptoms listed below and dosing should be reduced for intolerance (10):
• Nausea, vomiting
• Abdominal cramps
• Diarrhea
• Constipation
• Childhood poisoning. Note: Maternal iron preparations are the second most common cause of childhood poisoning in the United States (after aspirin).
Folate Deficiency Anemia
Background
• Folic acid deficiency is the most common cause of megaloblastic anemia in the United States.
• It complicates 1% to 4% of pregnancies in the United States and approximately 30% worldwide (2,10).
• Folate requirement increases from 50 μg/day in the nonpregnant state to 400 μg/day during pregnancy.
• Folate is a component of tetrahydrofolate (THF), which plays an important role in nucleic acid synthesis and maturation.
• Folate is solely obtained from the diet and absorbed in the proximal jejunum. Prime dietary sources are leafy vegetables, legumes, and animal protein.
• Similar predisposing factors to IDA in pregnancy, along with other specific considerations:
• Ongoing hemolysis (e.g., hemoglobinopathies)
• Seizure disorders treated with anticonvulsants, which interfere with metabolism
• Chronic disease promoting malabsorption, e.g., gluten-induced enteropathy and tropical sprue
• Likely confounded with socioeconomic or nutrition status, folic acid deficiency has been associated with such pregnancy complications as birth defects, low birth weight, abruptio placentae, and prematurity (21).
Etiology
• Folate stores are limited and easily depleted within 3 to 4 months in times of increased demand (e.g., pregnancy and lactation) (2).
• Low levels of folic acid result in disruption of nucleic acid synthesis, cellular division impairment, and accumulation of homocysteine.
Evaluation
History and Physical
• Deficiencies in either folic acid or B12 can present with hyperpigmentation, low-grade fever, glossitis, skin changes, and other nonspecific symptoms of anemia.
• However, the concomitant presence of neurologic symptoms is diagnostic of B12 deficiency and is almost never seen with folic acid deficiency.
Laboratory Tests
• Macrocytic anemia and megaloblasts present in the bone marrow; MCV is usually greater than 110 fL.
• The macrocytosis can be masked by concomitant iron deficiency or thalassemia (2% to 5%).
• Peripheral smear may demonstrate neutropenia, thrombocytopenia, and hypersegmented granulocytes.
• Serum folate levels are decreased to less than 2.0 μg/L (normal 2.0 to 15 μg/L).
• Red cell folate concentration (an indicator of severity of deficiency) is usually less than 160 μg/L (normal 160 to 640 μg/L).
• Plasma homocysteine and methylmalonic acid levels will differentiate between folate deficiency and vitamin B12 deficiency. Methylmalonic acid is normal in folate deficiency.
Treatment
Medications
• The recommended daily dose of folic acid is 1 mg for uncomplicated pregnancies.
• With treatment, an increased reticulocyte count should be seen within 3 to 4 days.
• If neurologic symptoms are present, a B12 level should be measured. Folic acid will correct the anemia, but not the neurological symptoms.
• Oral folic acid is sufficient for treatment. If folic acid antagonists are being used, parenteral folic acid is indicated.
Patient Education
• Prevention or reduction of neural tube defects
• Folic acid can be used preconceptionally for the prevention (or reduction) of both firsttime and recurrent neural tube defects (see Chapter 30, Teratogens and Birth Defects).
B12 Deficiency Anemia
Background
Etiology
• Vitamin B12 is readily available in conventional diets, and deficiency is rarely due to inadequate ingestion, except in strict vegetarians.
• Intrinsic factor is important to absorption and is produced by the fundic parietal cells.
• The same cells are responsible for hydrochloric acid secretion, which frees cobalamin (B12) from the ingested animal protein.
• Freed cobalamin is bound to intrinsic factor and absorbed in the ileum.
• Inadequate synthesis and production of intrinsic factor or malabsorption syndromes are the most common causes, as a result of these conditions listed below (1):
• Autoimmune inhibition of intrinsic factor production a.k.a. pernicious anemia (rare in this age group) and Zollinger-Ellison syndrome
• Previous gastric or intestinal surgery (i.e., gastric bypass and ileal resection)
• Intestinal parasites, HIV
• Atrophic gastritis, severe pancreatitis, or inflammatory bowel disease (Crohn disease)
• Medications (i.e., antihistamines and proton pump inhibitors)
• Neurological symptoms are related to damage to the posterior column of the spinal cord.
Evaluation
Laboratory Tests
• A radioimmunoassay is used to measure B12 serum levels.
• Vitamin B12 levels may fall to 80 to 120 pg/mL during pregnancy; levels below 50 pg/mL are indicative of B12 deficiency.
• Low levels of vitamin B12, but normal levels of folate are expected
• The Schilling test is used to measure B12 absorption but is contraindicated in pregnancy secondary to use of radioactive cobalt.
• However, if vitamin B12 deficiency is present, a Schilling test must be done at a safe time after delivery to rule out pernicious anemia.
• Elevated serum bilirubin and lactic dehydrogenase levels may also be demonstrated (2).
Treatment
Medications
• Treatment is vitamin B12 1 mg intramuscular daily for 1 week, then every week for 4 to 8 weeks, and continues at monthly intervals indefinitely.
• Serum B12 levels should respond within 6 weeks. Reticulocytes should increase within 3 to 5 days.
Anemia of Chronic Illness
Background
• Intestinal disease (e.g., peptic ulcer disease)
• Parasitic disease (e.g., malaria, helminthes)
• Chronic or subclinical infection (e.g., chronic renal or liver disease)
• Chronic inflammatory process (e.g., rheumatoid arthritis)
• Neoplasia
• Human immunodeficiency virus
Evaluation
Laboratory Tests
• Normochromic, normocytic (or microcytic) anemia usually unresponsive to iron therapy.
• The diagnosis of an underlying condition requires a high index of suspicion followed by careful history and physical examination.
• Treatment is guided by the specific cause.
APLASTIC ANEMIA
Background
• Aplastic anemia in pregnancy has widely varying reported outcomes.
• Pregnancy may be a causative factor in pre-existing aplastic anemia. Potential pathologic mechanisms suggested are hormonal inhibition of erythropoiesis or the bone marrow’s inability to respond to normal stressors of pregnancy.
• Conflicting evidence is termination of pregnancy only promoted recovery in one-third of the cases (22).
• The two most common causes of death in patients with aplastic anemia are infection and hemorrhage (23).
Etiology
• Aplastic anemia is defined as pancytopenia with a hypocellular bone marrow and no increase in reticulocytosis, in the absence of malignancy or myeloproliferative disease.
• Seventy percent to eighty percent of cases are idiopathic. The remaining have been associated with a multitude of causes, including medications, solvents (e.g., benzene), agricultural pesticides/fertilizers, and recreational drugs (e.g., MDMA, ecstasy) (22).
Evaluation
• History should be detailed and evaluate for recent medication use, potential occupational exposure, and family history for inherited causes. Fanconi anemia is most commonly associated; however, patients with history of paroxysmal nocturnal hemoglobinuria (PNH) and myelodysplastic syndrome may also share genetic properties with aplastic anemia (2).
• Clinical presentations include (23):
• Profound pallor
• Petechiae
• Bleeding
• Epistaxis
• Infection
Laboratory Test
• Pancytopenia is found on the CBC. Severe cases demonstrate ANC less than 500, platelets less than 20,000, and anemia with a retic count less than 1%.
• Bone marrow aspirate and biopsy are necessary to make the diagnosis (cellularity less than 25%) and to exclude other causes for pancytopenia (24).
Treatment
• Bone marrow transplant (BMT) with an HLA-matched donor is first-line treatment.
• High dose immunosuppressive therapy used in conjunction with the transplant makes BMT relatively contraindicated in pregnancy (22).
• Alternative immunosuppressive agents include androgen therapy, antithymocyte globulin (ATG), steroids such as prednisone, or monoclonal antibody therapy directed against helper T cells.
• Androgen therapy is contraindicated especially with female fetuses.
• ATG therapy is not widely used in pregnancy.
• Monoclonal antibody therapy, that is, cyclosporine, in adjunction with a granulocyte– macrophage colony stimulator has demonstrated no fetal toxicity and favorable pregnancy outcomes.
• Supportive care remains the mainstay of treatment in pregnancy and includes serial blood and platelet transfusions, treatment of infection, and stimulation of hematopoiesis with steroids (22,25).
• Recommendation for pregnancy termination remains uncertain unless maternal condition worsens.
• Maternal and fetal mortality rates depend on severity of disease and resource availability.
PAROXYSMAL NOCTURNAL HEMOGLOBINURIA
Background
Etiology
• Disorder results from an acquired somatic mutation of the PIG-A gene, which encodes for glycosyltransferase enzyme, on chromosome X.
• This key enzyme makes glycophosphatidylinositol (GPI), which is essential for transmembrane formation in anchoring CD55 and CD59, two integral regulatory proteins in complement activation.
Epidemiology
• A rare disorder with one to two cases per million individuals
• Twenty-five percent of cases discovered during pregnancy, although limited information available (1,2).
Diagnosis
Clinical Manifestations
• Clinical manifestations due to susceptibility of intravascular hemolysis:
• Hemolysis
• Hematuria (night hemolysis lead to morning tea-colored urine)
• Thrombosis
• Bone marrow failure
• Pregnant women experience severe anemia, preterm delivery, thrombocytopenia, low birth weight, and neonatal and maternal death.
• Twenty percent maternal mortality depending on severity of the enzyme deficiency (2)
• Thrombosis is the leading cause of death in PNH.
• Anticoagulation with low molecular weight heparin (LMWH) until 6 weeks postpartum is advised.
• Lab findings include severe anemia with low reticulocyte count.
• Fifty percent present with pancytopenia consistent with aplastic anemia (22)
Treatment
• Eculizumab, a monoclonal antibody therapy against complement 5, has demonstrated improvement in hemolysis, hemoglobin levels, and transfusion requirements (1).
• Early detection of infection is paramount.
• Transfusion support as needed.
Inherited causes of anemia
• Hemoglobinopathies
• Inherited hemolytic anemia
HEMOGLOBINOPATHIES
Background
• Normal hemoglobin is a tetramer with two sets of paired alpha (α), beta (β), delta (δ), or gamma (γ) chains.
• α-Globin gene pair is located on chromosome 16, along with the embryonic zeta (ζ) globin gene.
• β-globin gene is located on chromosome 11, along with the embryonic epsilon (ε) globin, two fetal γ-globins, and a δ-globin gene.
• The principle hemoglobin in adults, namely hemoglobin A1, is composed of two α chains and two β-chains and constitutes 97% of adult hemoglobin (2).
• Hemoglobin A2, which is normally less than 2% or 3% of the total adult hemoglobin, is composed of two α-chains and two δ-chains.
• Fetal hemoglobin F, two α-chains and two γ-chains, produced after 6 weeks postconception, predominates between 12 and 24 weeks’ gestation and is slowly replaced by A2 by a few weeks following delivery.
• In normal adults, hemoglobin F accounts for less than 1% of total hemoglobin.
• Embryonic hemoglobin, Hgb Gower consists of two ε and two ζ chains. It is not found in the fetus after the first trimester or in adults.
• Hemoglobinopathies are inherited single-gene disorders and affect the globin chains of the hemoglobin tetramer. Comprised of two main classes:
• Quantitative disorders: Thalassemia syndrome, which is characterized by inadequate production of structural normal hemoglobin, is named according to the hemoglobin affected.
• Structural disorders: Sickle cell disease characterized by a change in the amino acid configuration of the hemoglobin leading to its dysfunction (26).
Alpha-thalassemia
Background
• Commonly found in Southeast Asian, African, and Mediterranean populations
Etiology
• Manifestation of gene deletions in the α-globin chain genes.
• May inherit in conjunction with β-globin disorders and portends less severity in Hb SS or Hb SA.
• The clinical severity is based on number of alleles deleted.
• The genotype of normal individuals is aa/aa.
• The carrier state is represented by −a/aa and is clinically silent.
• α-Thalassemia minor: deletion in two of four copies of α-globin. Trans deletion (−a/−a) is more common in African descent and cis deletion ( —/aa) is common in Asians.
• Hemoglobin H disease is seen with a deletion of three α-chains (—/−a).
• Hb Bart is the absence of all four α-chains (−−/−−) and is incompatible with life.
• Less common, a mutation results in a normal α-globin gene
• Hb Constant Spring: long unstable α-chain due to stop codon mutation
• Hb Quong Sze: substitution impairing αβ dimer
• αTSaudi: point substitution likely affecting stability or translation of mRNA
Evaluation
• History and physical and laboratory findings all determined by severity (27)
• α-Thalassemia minor is usually clinically asymptomatic, except during great stress. It is usually an incidental finding in pregnancy.
 A mild microcytic, hypochromic anemia with poikilocytosis and anisocytosis is sometimes seen.
A mild microcytic, hypochromic anemia with poikilocytosis and anisocytosis is sometimes seen.
 Evidence of iron deficiency may develop in pregnancy.
Evidence of iron deficiency may develop in pregnancy.
• Hemoglobin H disease
 Occurs mostly in Southeast Asians
Occurs mostly in Southeast Asians
 Chronic moderate hemolytic anemia is present with reticulocytosis, microcytosis, hypochromasia, and poikilocytosis.
Chronic moderate hemolytic anemia is present with reticulocytosis, microcytosis, hypochromasia, and poikilocytosis.
 Splenomegaly and occasionally hepatomegaly is found along with hemochromatosis.
Splenomegaly and occasionally hepatomegaly is found along with hemochromatosis.
 Hb H (a tetramer of β-chains) and Hb Bart are both evident.
Hb H (a tetramer of β-chains) and Hb Bart are both evident.
– Newborns carry 2% to 10% of Bart’s hemoglobin (an abnormal tetramer of γ-chains).
• Hemoglobin Bart
 The fetus will generally experience second-trimester hydrops and result in intrauterine death.
The fetus will generally experience second-trimester hydrops and result in intrauterine death.
 Bart’s hemoglobin predominates
Bart’s hemoglobin predominates
 These pregnancies are notable for a high incidence of preeclampsia (2).
These pregnancies are notable for a high incidence of preeclampsia (2).
 Unusual in those of African descent
Unusual in those of African descent
• Couples at risk should be identified early and offered genetic screening.
• Carrier state may only be identified by molecular genetic testing.
• In those found to be carriers, genetic counseling should be encouraged for inheritance risk determination (28).
• Prenatal diagnosis with CVS, amniocentesis, and cordocentesis are all options, if mutation or deletion is previously identified in the parents.
• Free fetal DNA will become more widely available to evaluate for hemoglobinopathies, at this time approximately 65% are detected (29).
• Prior to pregnancy, preimplantation genetic diagnosis is also available.
Treatment
Medications
• In α-thalassemia minor, folate may need to be supplemented.
• α-Thalassemia carriers and minors usually do not behave different from normal gestations.
• In hemoglobin H disease, transfusions may be needed for severe anemia; otherwise, limited evidence is available regarding pregnancy outcome.
Beta-thalassemia
Background
• Similar ethnic, Mediterranean and African, predisposition as α-thalassemia.
• Mutation in β-globin chain gene leads to variable or no production of Hb A; this promotes red cell weakness and manifests as a chronic hemolytic anemia.
• Ineffective erythropoiesis encourages hemolysis, and extramedullary hematopoiesis leads to hemochromatosis.
Etiology
• β-Thalassemia minor is the heterozygous state.
• β-Thalassemia major is the homozygous state.
• Cooley’s anemia (severe form)
• β-Thalassemia intermedia
• Pregnancy rarely occurs because of delayed pubertal growth and anovulation.
Evaluation
• Laboratory findings of moderate to severe microcytic, hypochromic anemia (low MCV) with a relatively high red cell count and target cell morphology
• Hemoglobin A2 is elevated above 3.5% (usually 3.5% to 7%) and Hb F slightly elevated.
• Serum iron and ferritin concentrations are elevated.
• The clinical manifestations are variable in β-thalassemia minor.
• Thrombotic events complicate more pregnancies in those affected by β-thalassemia major and intermedia.
• Genetic testing performed is DNA analysis with PCR.
• Iron overload may be present from prior transfusions.
Treatment
• Folic acid supplementation may be necessary.
• Transfusions therapy in severe anemia is usually performed with aggressive iron chelation therapy (i.e., deferoxamine) to prevent iron accumulation.
• Deferoxamine has a teratogenic potential but has been safely used in the second and third trimester without fetal compromise.
Complications
• Infants who survive with this anemia have a course that is marked by profound, transfusiondependent anemia, growth delay, delayed sexual development, and heart failure by puberty.
• Pregnant women with evidence of cardiac impairment from iron overload have less favorable outcomes and high maternal morbidity/mortality.
• Pregnancies also experience higher rates of preterm delivery, third-trimester intrauterine death, and growth restriction.
Sickle Cell Anemia
Background
• Autosomal recessive disorder originating from a point mutation, which results in a β-chain substitution and production of defective hemoglobin.
• Severity of disorder influenced by zygosity of inheritance.
• Best perinatal outcomes in prenatal care coordinated by multidisciplinary team to include the obstetrician, hematologist, and anesthesiologist.
• Since 1 in 500 births in the African American population is affected by Hb SS, sickle cell disease is now part of the universal newborn screening in the United States (2).
Etiology
• A single nucleotide change (alanine → thymine) in the coding sequence creates a substitution of glutamic acid to valine. Subsequent production of HbS has the following manifestations:
• In a setting of low oxygen tension, RBCs become sickle shaped with decreased elasticity and increased fragility.
 Microvascular obstruction with rigid RBCs leads to vasoocclusive crises in long bones, chest, back, and abdomen.
Microvascular obstruction with rigid RBCs leads to vasoocclusive crises in long bones, chest, back, and abdomen.
 Increase in adhesion and coagulation factors also encourages RBC adhesion and platelet aggregation worsening the vasoocclusive cycle.
Increase in adhesion and coagulation factors also encourages RBC adhesion and platelet aggregation worsening the vasoocclusive cycle.
 Normal physiologic changes of pregnancy promote more sickling especially in the later gestation. Sickling in placental vessels and intervillous space may be associated with adverse perinatal events.
Normal physiologic changes of pregnancy promote more sickling especially in the later gestation. Sickling in placental vessels and intervillous space may be associated with adverse perinatal events.
• Shortened life span of RBCs (protective in malaria endemic countries)
 Skeletal adaptations occur secondary to marrow cavity expansion.
Skeletal adaptations occur secondary to marrow cavity expansion.
• Inability to concentrate urine (hyposthenuria) may occur as a result of renal papillary necrosis and loss of deep medullary nephrons.
• By adolescence, functional asplenia has occurred as a result of repetitive microinfarctions. Therefore, increased risk of infections with encapsulated organisms (i.e., Streptococcus. pneumonia, Haemophilus. flu, meningococcus).
• Other single amino acid substitutions can cause defective hemoglobin that lead to rigid RBCs with reduced solubility and a changed oxygen affinity.
• Hb C, a point mutation of alanine > guanine, substitutes adenine for glutamic acid.
• Hb Sb thalassemia (discussed in previous section).
Diagnosis
• The course of Hgb S disease prior to pregnancy frequently predicts how the woman will do in pregnancy. Avoid hypovolemia and hypoxemia in any trimester (2)
• Genetic counseling and prenatal diagnosis should be offered (28,29)
• Hemoglobin electrophoresis is the diagnostic test.
• Moderate anemia with irreversibly sickled RBCs on peripheral smear. Normal, target, fragmented, and nucleated cells may also be seen.
• Hemoglobin electrophoresis shows greater than 80% hemoglobin S and elevated Hgb F.
• Solubility test (e.g., Sickledex) is inadequate for distinguishing between genotypes.
Clinical Manifestations
• β-Chain synthesis does not reach sufficient levels to cause symptoms until about 6 months of age.
• Children with Hgb SS are at increased risk for certain infections:
• Sepsis
• Meningitis
• Pneumonia
• Osteomyelitis
• Urinary tract infections
• Respiratory manifestations are secondary to vasoocclusion or infection.
• Acute chest syndrome: pulmonary infiltrate, fever, pleuritic chest pain, and tachypnea. Treat if infectious, otherwise supportive care with hydration, pain relief, adequate oxygen.
• Leading cause of death is from acute chest syndrome or pulmonary embolism.
• Cardiovascular manifestations are secondary to the condition’s hyperdynamic state.
• Cardiomegaly (50% with left ventricular hypertrophy)
• Prolonged PR interval
• Increased risk for gestational or worsening pulmonary hypertension, preeclampsia, and intrauterine growth restriction (IUGR)
• Frequent complications in pregnancy: (30)
• Antepartum admission
• Thrombotic events
• Placental abruption
• Vasoocclusive episodes
• Postpartum infections
• Vasoocclusive target areas:
• Extremities
• Lungs
• Spleen, splanchnic bed
• Kidneys
• Brain (CVA, moyamoya)
• Eyes (retinopathy)
• Perinatal complications include small-for-gestational age (SGA) neonate, preterm labor, and premature rupture of membranes (30) In efforts to decrease the likelihood of developing pre-eclampsia, a daily lose-dose (81 mg) aspirin may be considered after 12 weeks gestation.
Treatment
• Folic acid supplementation of 5 mg/day is recommended.
• Iron supplementation is not helpful in treating anemia. Most patients have iron overload due to transfusion history (26).
• Sickle cell crises must be treated with generous fluid support, oxygen supplementation, pain management, and blood transfusions as indicated.
• Hydroxyurea is an antineoplastic agent that encourages production of hemoglobin, and decreases the severity of sickling and frequency of pain crises. Concerns for fetal toxicity, so controversial in pregnancy.
• Blood transfusion to treat vasoocclusive crises and anemia:
• Partial exchange transfusion targets a concentration of hemoglobin A greater than 40% to 50% and a hematocrit between 25% and 30%.
• Evidence has shown a decrease in number of vasoocclusive crises, but no difference in perinatal outcome (31).
• Prophylactic transfusions are not recommended but may be performed in setting of severe anemia (Hgb less than 6), acute chest syndrome, preoperative, sepsis, acute renal failure, or protracted pain crisis.
• Transfusion therapy risks include 25% rate of alloimmunization, 20% rate of delayed transfusion reaction, iron overload, and 1:2,000,000 risk of HIV (2,31).
Procedures
• Careful monitoring for asymptomatic bacteriuria and urinary tract infections is necessary.
• Fetal surveillance should be started in the late second to early third trimester with nonstress tests and sonographic evaluation for growth and amniotic fluid index (AFI).
• Labor and delivery are managed on the basis of obstetric principles.
• Cesarean section should be performed only for obstetric indications. Regional anesthesia is preferred to general anesthesia.
• Venous thromboembolism (VTE) prophylaxis postpartum is recommended.
• Influenza vaccine should be within a year and penicillin prophylaxis continued. Administer pneumococcal vaccine every 5 years.
Patient Education
• Limited evidence of good quality to advise one contraception method over another; however, depot medroxyprogesterone acetate has been suggested to decrease the incidence and severity of vasoocclusive crises. No studies evaluate the risk of thromboembolism with combined hormonal contraception (32).
• Permanent sterilization should be considered when the patient has completed childbearing.
• Termination of the pregnancy because of maternal sickle cell anemia is largely unwarranted and should be considered on an individual basis.
Sickle Cell Trait (Hb SA)
Evaluation
• Carriers will be heterozygous for the condition and have one abnormal and one normal β-chain (1:12 African Americans) on Hgb electrophoresis.
• Family history is important (28)
Clinical Manifestations
• Sickle cell trait is associated with increased rates of pyelonephritis in pregnancy (26).
• In one study, carriers had a higher incidence of pyelonephritis despite similar rates of asymptomatic bacteria or acute cystitis compared to normal pregnant women.
• Urinalysis should be performed at each visit with urine cultures every trimester.
• Patients at increased risk of early spontaneous abortion and multiple gestations but appears to have low risk of preterm delivery at less than 32 weeks (30).
• In settings of significant hypoxemia, sickle cell trait may be associated with:
• Vasoocclusive disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


