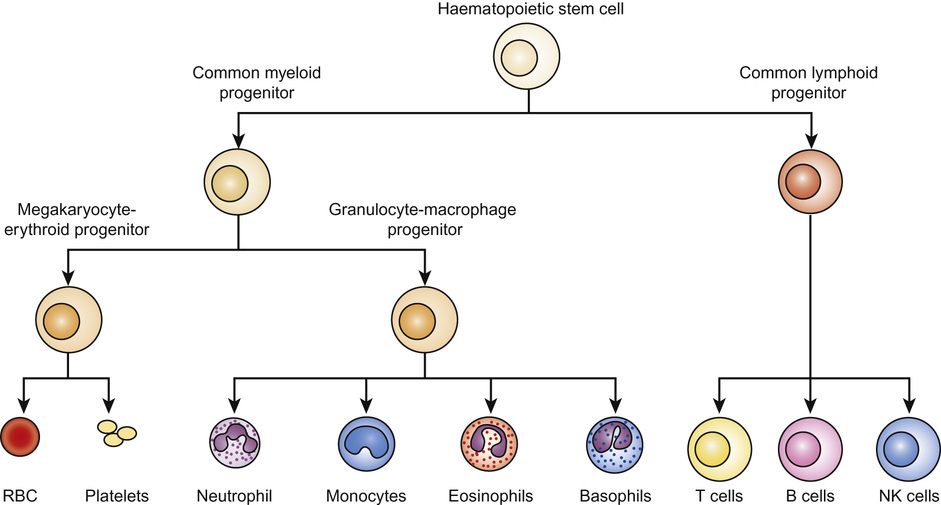
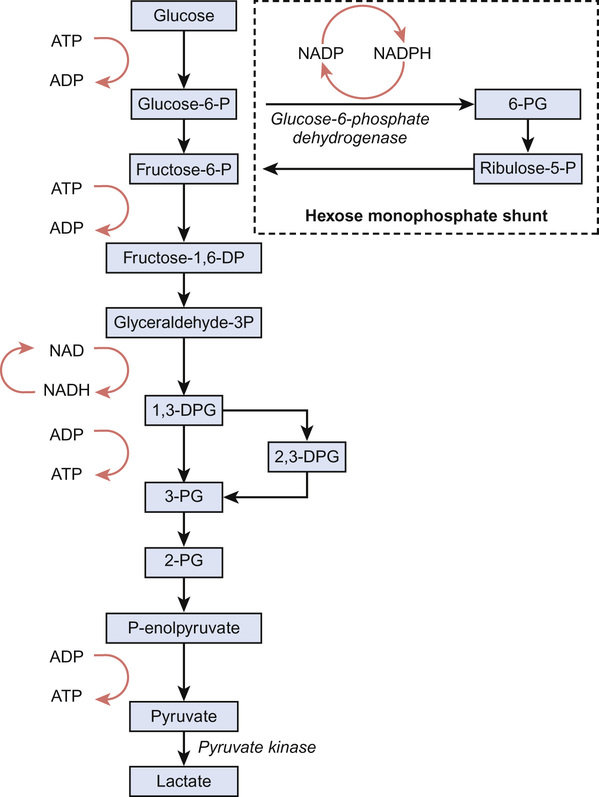
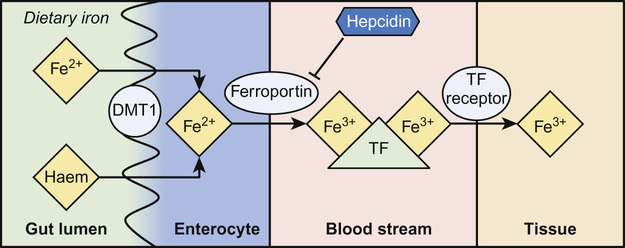
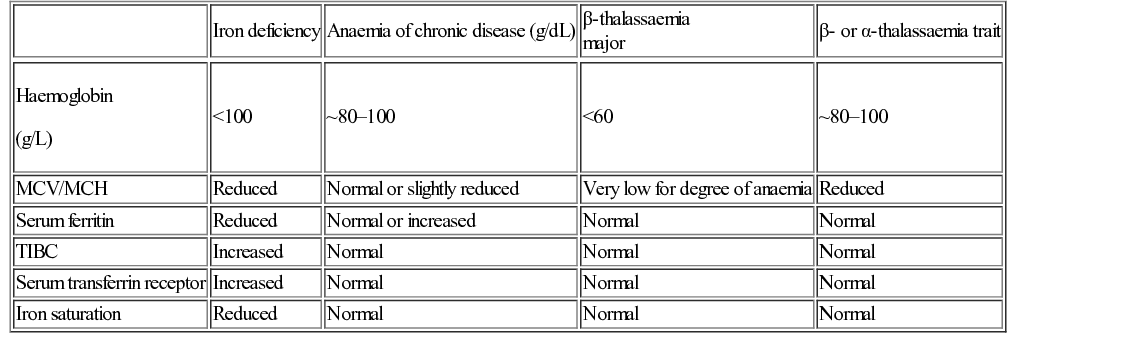
Irene A G Roberts, David O’Connor • Understand haematopoiesis in children • Recognize the genetic and environmental factors involved in haematological disorders • Understand the investigation and management of disorders of haematopoiesis • Understand the normal coagulation pathway and its investigations • Understand the pathophysiology, investigation and management of disorders of haemostasis/thrombosis Haematopoiesis is the process through which all types of mature blood cell are produced. Haematopoietic stem cells (HSCs) are rare, multipotent cells characterized by their ability to both ‘self renew’ (proliferate) and to mature into fully differentiated cells of any of the haematopoietic lineages (Fig. 23.1). HSCs sustain blood cell production throughout life. The principal haematopoietic lineages are: the erythroid/megakaryocytic lineage, which gives rise to red cells and platelets; the granulocyte/macrophage lineage, which gives rise to granulocytes and monocytes; and the lymphoid lineage, which gives rise to B cells, T cells and NK cells. Amplification in cell numbers occurs as HSCs differentiate and it is estimated that each HSC can produce ~106 cells after undergoing up to 20 cell divisions. Disruption of stem cell function can result in a variety of blood disorders, including leukaemia (see Chapter 22, Oncology) and aplastic anaemia. HSCs can be characterized on proteins expressed on the cell membrane, such as CD34. These cell markers can also be used to purify HSC for clinical applications, including haematopoietic stem cell transplantation (HSCT). The regulation of haematopoiesis is complex. HSCs and their progeny are controlled by a network of interactions with haematopoietic growth factors and cellular components of the haematopoietic microenvironment (e.g. stromal cells) that maintain balanced blood cell production. Similar regulatory mechanisms respond to increased demand for specific blood cell types, such as increased red cell production in anaemia or increased granulocyte production in infection. Primitive haematopoiesis begins in the yolk sac during the first few weeks of embryonic life and gives rise mainly to red blood cells. Primitive haematopoiesis is transient and is replaced at 5–6 weeks’ gestation by definitive haematopoiesis, which has the capacity to produce all blood cell types. Definitive HSCs develop in the aorta-gonad-mesonephros (AGM) region of the dorsal aorta. HSCs migrate from the AGM to the fetal liver and spleen from 6–7 weeks’ gestation. The liver then forms the primary site of haematopoiesis throughout fetal development. During the third trimester, haematopoiesis progressively increases in the bone marrow so that this becomes the main site of haematopoiesis shortly before birth. Initially, haematopoiesis occurs in all areas of bone marrow, becoming restricted to the axial skeleton and the proximal ends of the long bones later in childhood. Red cells are specialized cells that mainly function to deliver oxygen to the tissues and to remove carbon dioxide. They are biconcave (disc-shaped), lack a nucleus and contain large amounts of the oxygen-carrying protein haemoglobin (Hb). Each molecule of Hb consists of four globin chains and a central iron-containing haem group. The composition of Hb changes in an ordered sequence during fetal development (Table 23.1). The first globin chains produced are epsilon (ε)- and zeta (ζ)-globin, followed almost immediately by gamma (γ)-globin chains, which together give rise to two types of embryonic Hb. Alpha globin synthesis begins shortly after this so that HbF (α2γ2) is produced from 3–4 weeks of fetal life and is the predominant Hb until after birth. Adult Hb (HbA: α2β2) remains at low levels (10–15%) until 30–32 weeks’ gestation. After this, the rate of HbA production increases at the same time as HbF production falls. As a result, the average HbF level at birth in term babies is 70–80%, with a HbA of 25–30%, small amounts of HbA2 and sometimes a trace of Hb Barts (γ4). After birth, HbF falls rapidly (to ~2% at age 12 months) with a corresponding increase in HbA. In term babies there is little change in HbF in the first 15 days after birth. In preterm babies who are not transfused, HbF may remain at the same level for the first 6 weeks of life before HbA production starts to increase. This delay in HbA production (i.e. the switch from the γ-globin of HbF to the β-globin of HbA) makes the diagnosis of some haemoglobinopathies (see below) difficult in neonates. The major function of Hb is to deliver oxygen to tissues. The binding and release of oxygen causes small changes to the configuration of globin chains in the Hb molecule, which in turn alters the affinity of Hb for oxygen. As oxygen is unloaded, the Hb molecule opens up allowing 2,3-diphosphoglycerate (2,3-DPG) to enter, reducing oxygen affinity and ensuring that the Hb molecule does not take up oxygen from the tissues. These changes in affinity result in the typical sigmoid shape of the oxygen dissociation curve, which is discussed in more detail in Chapter 17, Respiratory medicine. To function effectively, red cells must be able to generate energy (in the form of ATP) and reduce molecules to prevent oxidative damage. The Embden–Meyerhof pathway metabolizes glucose to lactate, producing two molecules of ATP, which are used to maintain red cell shape and osmotic gradient (Fig. 23.2). In addition, this pathway generates NADH (which is used to reduce iron to the active ferrous [Fe2+] form) as well as 2,3-DPG. The pentose phosphate pathway is an alternative, essential glycolytic pathway, which generates NADPH, another important source of reducing power that maintains iron in the active form. Deficiency of glucose-6-phosphate dehydrogenase (G6PD), a key enzyme in the pathway, results in marked susceptibility to oxidative stress. The average red cell lifespan is 120 days in older children but ~90 days in neonates, during which time each red cell is estimated to travel 300 miles through the circulation. To prevent damage to red cells and maximize their flexibility, the specialized red cell membrane consists of a complex lipid bilayer containing a number of key scaffolding proteins. The most abundant protein is spectrin, which forms tetramers and attaches to the membrane via the ankyrin protein. Abnormalities in these proteins cause membrane disorders such as hereditary spherocytosis and hereditary elliptocytosis. Dietary iron is vital for erythropoiesis. Iron deficiency due to inadequate intake or chronic blood loss is the most common cause of anaemia worldwide. Foods rich in iron include meat (especially liver), nuts and pulses. Several of the genes which encode iron regulatory proteins have been identified. This has led to better understanding of iron metabolism in humans and to the identification of the genetic basis of several inherited iron overload and iron deficiency disorders. Iron absorption occurs in the duodenum: • Iron in the diet is absorbed by enterocytes either in the Fe2+ form, via the divalent metal transporter (DMT)1 receptor (Fig. 23.3), or as haem. The DMT1 receptor and the transferrin receptor 1 (TfR1) are both important regulators of body iron. The synthesis of these proteins is controlled by iron regulatory proteins (IRPs) that can bind to iron response elements (IREs) on genes. When iron levels are adequate, IRPs do not bind, which allows production of ferritin and therefore storage of iron. When iron deficiency occurs, IRPs bind to the IREs, blocking production of ferritin and enhancing synthesis of DMT1 and TfR1, which encourages increased iron absorption in the gut and uptake by the tissues. Mutations in DMT1 cause congenital anaemia. Mutations in the ferroportin gene are associated with congenital hyperferritinaemia and iron overload. Iron is present in the body in several forms. The greatest amount of iron is in haemoglobin, which can be recycled once red cells reach the end of their life. The other main form of iron storage is ferritin, a water-soluble molecule found in many tissues. The serum ferritin concentration provides a fairly accurate estimation of total body iron levels (in the absence of inflammation) and is a useful investigation when iron deficiency is suspected. Iron is also present in an insoluble form called haemosiderin, in myoglobin in the muscles and in many important enzymes. In general, the amount of iron in the diet is excess to requirements and so typically only 5–10% of dietary iron is absorbed. However, during periods of increased need, such as iron deficiency, iron absorption can be increased to up to 30%. Iron availability is very tightly regulated at both the cellular and systemic levels in order to avoid iron deficiency or iron overload. Key to this is hepcidin, a recently discovered small peptide hormone produced by the liver, which plays a major role in controlling iron flux to plasma from enterocytes and macrophages through degradation of the cellular iron exporter ferroportin (see Fig. 23.3). Under steady state conditions, normal hepcidin levels prevent excess iron absorption and iron overload. Similarly, in iron deficiency, hepcidin levels fall. Thus, the hepcidin–ferroportin axis is essential for the maintenance of iron homeostasis and mutations in the genes which encode proteins of the hepcidin-activating pathway are a major cause of iron overload. Hepcidin insufficiency and increased iron absorption are also characteristic of anaemia due to ineffective erythropoiesis. In this situation, hepcidin is suppressed by the high erythropoietic activity, despite high total body iron, thereby worsening both the iron overload and the anaemia. Similarly, hepcidin excess due to mutations in the hepcidin inhibitor, transmembrane protease serine 6 (TMPRSS6) leads to iron refractory iron deficiency. There is no mechanism for regulating iron excretion if iron levels are too high. Therefore, patients receiving regular blood transfusions, and some patients with severe haemolytic anaemia, can become iron overloaded. Excess iron is then deposited in various organs, including the liver and heart, which can result in serious damage and organ failure (see later in chapter). Anaemia is defined as a haemoglobin level below the normal range. The normal range varies with age. Hb values are higher in utero due mainly to the higher oxygen affinity of HbF and fall during the first few months of life due to reduced red cell production. Thereafter, Hb values stabilize until puberty when growth leads to an increased demand for iron and iron deficiency may develop, particularly in girls. A practical age-based definition of anaemia which takes these physiological factors into account is: Anaemia occurs through three main mechanisms: The investigation of anaemia should begin with a full blood count and a blood film. If anaemia is associated with abnormalities in the white cell and platelet counts, a bone marrow disorder, such as leukaemia, should be suspected. However, where the white cell and platelet counts are normal, the mean cell volume (MCV) and mean cell haemoglobin (MCH) provide the most useful information about the likely aetiology (Table 23.2). Iron deficiency results in a microcytic hypochromic anaemia (low MCV, low MCH). β-Thalassaemia is an important differential diagnosis, particularly in children from the Indian subcontinent. In β-thalassaemia carriers (β-thalassaemia trait), the MCH and MCV are also low but the Hb is only slightly reduced (80–100 g/L). Further tests should be performed to confirm iron deficiency and rule out less common causes, such as thalassaemias and anaemia of chronic disease (Table 23.3). Red cell aplasia causes anaemia due to reduced or absent red cell precursors in the bone marrow. There are three main causes in childhood: Diamond–Blackfan anaemia (DBA) is a rare (4–7 cases/million live births) genetic disorder that usually presents with anaemia at birth or during infancy. It may present in fetal life and occasionally in older children. DBA is associated with physical abnormalities in 50% of cases, including: • Craniofacial abnormalities (cleft palate, typical facies) • Thumb abnormalities (hypoplastic, triphalangeal) In most cases, inheritance is autosomal dominant and a family history is present in 10–20% cases. Genetic studies have identified mutations in various ribosomal protein genes (the most common mutation being in RPS19 which occurs in ~25% of cases). These studies indicate that defective ribosomal biosynthesis is the primary cause of DBA resulting in apoptosis and dysfunction in other key pathways. Ribosomes are vital for protein synthesis but their exact role in DBA is yet to be fully elucidated. Laboratory tests in DBA will show: When DBA is suspected, a bone marrow biopsy should be performed which will confirm the diagnosis by showing a reduction in erythroid precursors while other cell types are unaffected. Screening for ribosomal protein mutations is now available and can be useful to confirm the diagnosis. After diagnosis, most patients are commenced on oral prednisolone, which induces a response in 70% of cases. It is usual to defer steroids until after initial MMR vaccination has been carried out. In children who are refractory to steroids, or where steroids cause unacceptable side effects, blood transfusions form the mainstay of treatment. Around half of all patients with DBA are maintained on regular transfusions, which leads to transfusional iron overload in the long term. Recently, HSCT has become a curative option for patients with DBA, usually using a healthy sibling donor. The main differential diagnosis of DBA in neonates is parvovirus-induced red cell aplasia. The main differential diagnosis in infants and young children is transient erythroblastopenia of childhood (TEC). Although DBA usually presents in infancy and TEC has a median presentation of 2 years, overlap in age groups can occur. TEC is a rare (5 cases/million) transient red cell aplasia that is thought to be triggered by an unknown infective agent. It is important to differentiate TEC from DBA as recovery occurs spontaneously in TEC, usually within 4–8 weeks, and so usually no treatment is needed. In the absence of a family history or physical abnormalities, laboratory tests can help to establish the diagnosis (Table 23.4). Anaemia in TEC is usually normocytic and is associated with neutropenia in some cases. HbF and eADA are normal. Table 23.4 Differentiating Diamond–Blackfan anaemia (DBA) and transient erythroblastopenia of childhood (TEC) Haemolysis increases the breakdown of red blood cells. If the bone marrow is unable to compensate for the increased red cell turnover, anaemia develops. Haemolysis can be broadly grouped into inherited causes (usually due to intrinsic red blood cell abnormalities) and acquired causes (usually due to extrinsic abnormalities, such as antibody-mediated destruction in immune haemolytic anaemias; Table 23.5). In children, intrinsic red cell abnormalities account for the majority of cases. Blood tests show a characteristic profile in haemolytic anaemia: Haemolysis may occur in the circulation (intravascular) or in various organs, such as the spleen (extravascular). Intravascular haemolysis leads to depletion of haptoglobin, increased LDH and large numbers of fragmented red blood cells, called schistocytes. Haptoglobin binds free haemoglobin in plasma to form a haptoglobin–haemoglobin complex, which is then removed from the circulation by the reticuloendothelial system (mostly the spleen). After haptoglobin is depleted, free haemoglobin is filtered by the kidney. This reabsorption is overwhelmed in severe cases of intravascular haemolysis. LDH is present in many tissues but has high concentrations within red blood cells. Extravascular haemolysis usually takes place in the spleen or liver but can also occur in the lung. Haemolysis occurs when spleen and liver macrophage Fc receptors bind immunoglobulin attached to red blood cells and then either ingest small portions of the cell membrane creating spherocytes or phagocytose the whole cells. Amino acids from the globin chains are recycled and the iron is removed from the haem and reused. The haem is degraded into unconjugated bilirubin. In most cases the blood film provides useful clues to the diagnosis and aetiology, for example: • Sickle-shaped cells – diagnostic of sickle cell disorders • Spherocytes – seen in hereditary spherocytosis but also in immune haemolytic anaemias The direct antiglobulin test (DAT), which detects the presence of antibodies coating red blood cells, is essential for the diagnosis, or exclusion, of immune-mediated haemolytic anaemia. To perform the test, red blood cells are incubated with anti-human globulin (AHG) and if antibody is present on the cells, the AHG will cause agglutination. This agglutination is recorded as a positive DAT. Examples of conditions in children in which the DAT is positive are: • Haemolytic disease of the newborn (HDN) • Autoimmune haemolytic anaemia • Drug-induced haemolytic anaemia A 5-year-old girl with known hereditary spherocytosis (HS) presents with pallor and lethargy that has developed over the last 2 days. She is not jaundiced but she has a 5 cm palpable spleen and her Hb is 32 g/L. She had been very well until the previous week, when she developed signs of a mild coryzal illness, followed by increasing lassitude and anorexia. What is the pathogenesis of HS and how is it inherited? HS is the most common of a group of inherited disorders of the red cell membrane that also includes hereditary elliptocytosis. Most cases of HS are due to a mutation in the spectrin or ankyrin genes that encode for a key red cell membrane structural protein, which anchors the lipid membrane of the red cell to the protein skeleton. Loss of this anchoring effect allows small membrane fragments to be lost during red cell passage through the spleen. As membrane loss progresses, the red cells gradually become spherical. Eventually these spherocytic red cells are destroyed in the spleen resulting in haemolytic anaemia.
Haematology
Physiology of haematopoiesis
Development of haematopoiesis
Physiology of red blood cells
Haemoglobin
Red cell metabolism
The red cell membrane
Iron metabolism
Disorders of haematopoiesis
Anaemia
Investigation of anaemia
Red cell aplasia
Diamond–Blackfan anaemia
Transient erythroblastopenia of childhood
DBA
TEC
Physical abnormalities
50%
Absent
Family history
10–20%
Absent
MCV
High
Normal
Neutrophil count
Normal
Reduced in up to 50%
HbF
Increased
Normal
eADA
Increased
Normal
Mutations in ribosomal protein genes
Found in >50%
No
Spontaneous recovery
Occasional
Always
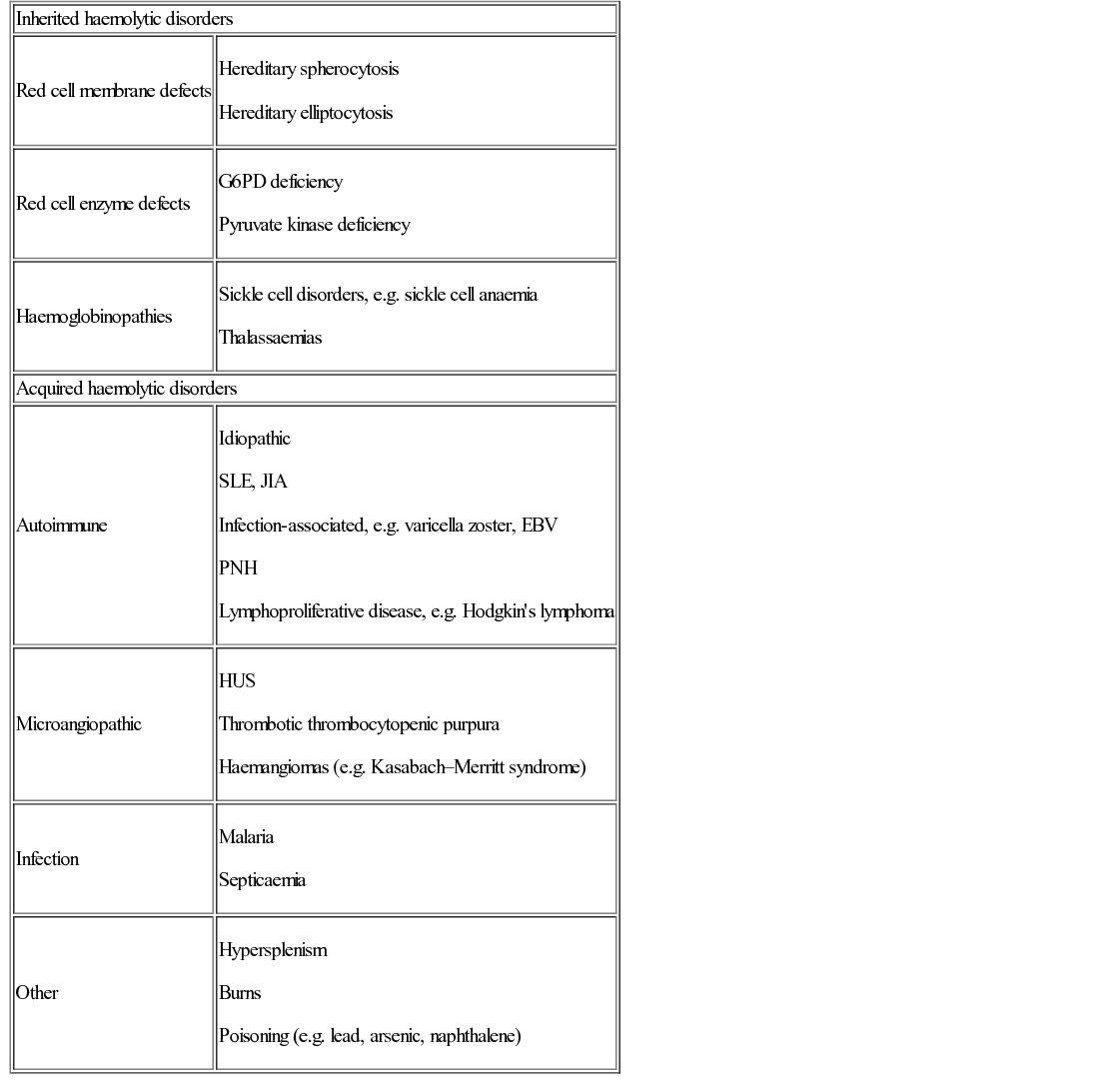
Increased red cell destruction (haemolysis)
Causes and mechanisms of haemolytic anaemia
The direct antiglobulin test
 Case history
Case history
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



Haematology
Chapter 23
Learning objectives
By the end of this chapter the reader should: