Glomerular Diseases
Allison A. Eddy
Glomerular diseases present clinically in several different ways, depending on the nature and severity of the primary disease and the extent to which the normal physiological functions of the glomerulus are perturbed.1,2 Some children with glomerulonephritis (GN) are found incidentally to have microscopic hematuria or proteinuria when checked by routine urinalysis but are otherwise asymptomatic. At the other extreme, children may become critically ill with oligoanuric rapidly progressive GN in need of urgent dialysis. A few glomerular diseases are inherited (see Chapter 473), but most forms of GN are acquired and are generally considered to be immunologically mediated. There are three classical clinical syndromes that develop from glomerular injury: acute and chronic glomerulonephritis, defined by the triad of hematuria, hypertension, and azotemia; nephrotic syndrome, defined by proteinuria and hypoalbuminemia; and hemolytic uremic syndrome, defined by microangiopathic hemolytic anemia, thrombocytopenia, and renal insufficiency.
GLOMERULONEPHRITIS
The pathophysiological sequence of events that lead to the development of the nephritic triad (hematuria, azotemia, and hypertension) are shown in Figure 472-1. In each of the clinical entities with glomerular proliferation, the inflammation process leads to decreased glomerular perfusion, resulting in compromised renal function and retention of salt and water with potential development of hypertension and edema.
APPROACH TO A PATIENT WITH GLOMERULONEPHRITIS
The patient with glomerular disease presents clinically with a constellation of features that may include hematuria, proteinuria, hypertension, edema, and renal insufficiency. The urinary sediment is characterized as active when dysmorphic erythrocytes and cellular casts are present. A series of questions guides the initial diagnostic and management plan.1
1. Does this patient have acute or chronic GN? Many patients with chronic GN appear relatively asymptomatic until the disease is advanced. Clues of chronicity include significant anemia, evidence of renal osteodystrophy (abnormal bone radiographs or an elevated PTH level), or small echogenic kidneys on ultrasound examination. Acute onset of severe hypertension often causes neurological symptoms such as headaches and seizures, while long-standing hypertension of insidious onset may be clinically silent, but left ventricular hypertrophy may be present.
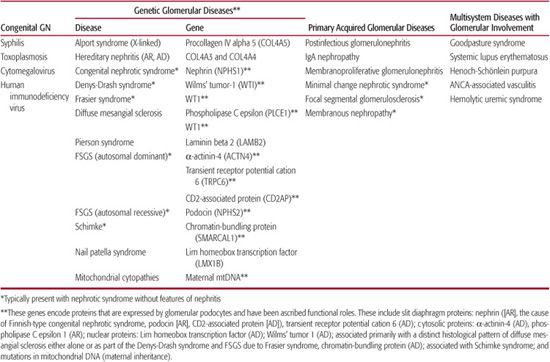
2. Does this patient have isolated renal disease, or are extrarenal organ systems involved? A careful systems review and physical examination will help determine whether the investigation should move in the direction of primary (acquired) GN or toward multisystem disease associated with GN (Table 472-1). Relevant extrarenal involvement may be clinically silent. For example, postinfectious serological studies (such as a streptozyme or anti–hepatitis B or anti–hepatitis C antibodies) may be indicated if infection-associated GN is a possibility. When patients present with vasculitis caused by Wegener’s granulomatosis, involvement of the lung parenchyma or sinuses usually requires radiological confirmation.
3. Does this patient have hypocomplementemic GN? A low serum C3 concentration generally indicates one of four diseases in children: acute postinfectious GN, lupus nephritis, membranoproliferative GN (MPGN), and GN associated with chronic infections (subacute bacterial endocarditis and shunt nephritis), as indicated in Figure 472-1.
4. Does this patient have recurrent painless macroscopic hematuria? If yes, IgA nephropathy is a likely diagnosis, although this also is a fairly common presentation of Alport syndrome during the first decade of life.
5. What is the age of the patient? Although most forms of GN can occur in almost any age group, most diseases have a characteristic age range for disease onset (Table 472-2). GN in the newborn period is extremely rare and is often the result of a congenital infection.
6. Does this patient have clinical evidence of rapidly progressive GN? Such a course is usually caused by crescentic GN and, more rarely, may result from acute GN with superimposed acute tubular necrosis. Rapidly progressive GN is an emergency that needs urgent histological diagnosis. Most of these patients have aggressive diseases that are successfully treated only with immunosuppressive therapy, if treatment is initiated early and in high doses. In patients with anti-GBM nephritis and severe vasculitis, early plasmapheresis may be lifesaving.
7. Does this patient have nephritic/nephrotic syndrome? Although proteinuria is a hallmark of significant glomerular disease, frank nephrotic syndrome is a less common feature of the acute nephritic syndrome. In a patient presenting clinically with acute GN and a low serum C3 level, the presence of nephrotic syndrome should suggest membranoproliferative GN or lupus nephritis. Nephrotic syndrome can occur in association with acute poststreptococcal GN, but this is rare.
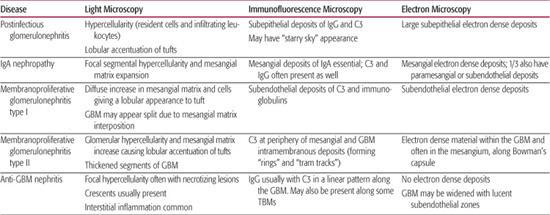
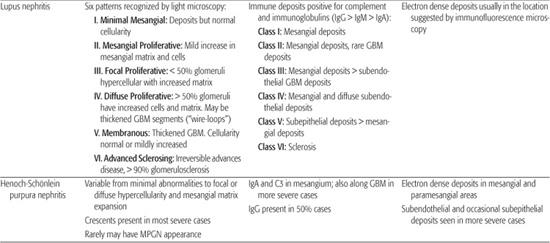
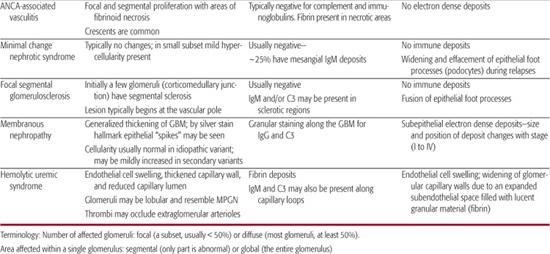
8. Does this patient need a renal biopsy now? Determining when to perform a renal biopsy is somewhat arbitrary. This procedure is generally not necessary when the diagnosis is obvious and the disease is self-limited without specific therapy (ie, typical acute poststreptococcal GN and mild nephritis secondary to Henoch-Schönlein purpura). For most glomerular diseases, a definitive diagnosis can be made only by renal biopsy. For many of these patients, the only way to design a rational and evidence-based plan of therapy is by histological confirmation of the diagnosis and the pattern and severity of renal damage (Table 472-3).
ACUTE POSTSTREPTOCOCCAL GLOMERULONEPHRITIS (APSGN)
APSGN is the most frequent and best-characterized acute postinfectious glomerulonephritis (GN). However, many other bacterial, viral, and parasitic pathogens may also induce postinfectious acute GN. Identifying a specific causative pathogen is often difficult, since the infection usually precedes nephritis by a few weeks. A history of pharyngitis or pyoderma suggests a previous infection by group A, β-hemolytic streptococcal disease that may be confirmed by serological testing. APSGN accounts for 80% to 90% of such cases and is used as the prototype for this group of disorders.
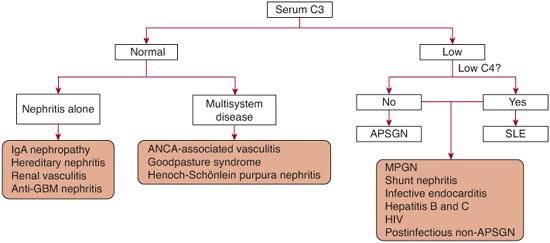
FIGURE 472-1. Approach to the evaluation of a child with glomerulonephritis, based on serum C3 level. ANCA, antineutrophil cytoplasmic antibody; ASPGN, acute poststreptococcal glomerulonephritis; HIV, human immunodeficiency virus; MPGN, membranoproliferative glomerulonephritis; SLE, systemic lupus erythematosus.
Table 472-1. Classification of Common Pediatric Glomerular Diseases
 EPIDEMIOLOGY
EPIDEMIOLOGY
APSGN is primarily a disease of school-age children (5 to 15 years; younger in epidemic forms) and is more common in boys. Although reported in an 8-month-old child, the disease is rare under 3 years of age and may occur as a sporadic or epidemic disease. APSGN follows infection with specific “nephritogenic” strains of group A β-hemolytic streptococcus. APSGN has been shown to be nephritogenic following pharyngitis (strains 1, 3, 4, 12, 18, 25, and 49) or impetigo (strains 2, 49, 55, 57, and 60).
Antibiotic treatment of the prodromal disease does not prevent acute GN, but treatment is important as a public health measure to prevent the spread of the nephritogenic bacteria. Although difficult to determine with certainty, the overall risk of developing APSGN after infection with a nephritogenic strain is in the range of 10% to 15%. The true incidence is difficult to determine, because 50% to 85% of patients with APSGN are asymptomatic. The incidence of this illness has decreased in the United States and Europe over the past two decades, likely due to earlier recognition and treatment of streptococcal infections. The prevalence of certain nephritogenic stains is decreasing as well. A seasonal pattern exists for APSGN in North America that mirrors that of the pathogenic organisms: pharyngitis in the winter and spring and impetigo in the summer and fall.
Table 472-2. Features of Common Pediatric Glomerular Diseases at Initial Presentation
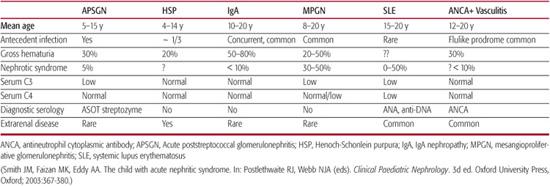
Table 472-3. Classical Histological Features of Pediatric Glomerular Diseases
 PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
The precise nature of the antigen-antibody complex that causes nephritis remains unclear.4 Although several streptococcal antigens have been identified in the glomerular immune deposits, two proteins are of particular interest cysteine proteinase exotoxin B (SpeB) and nephritis-associated plasma receptor (NAPlR) 
It is currently believed that target streptococcal antigen is initially trapped within glomeruli, with subsequent immune complex formation occurring in situ in the kidney. Once glomerular immune deposits are formed, the alternative and lectin complement pathways are activated, followed by neutrophil infiltration and glomerular damage. APSGN is an “exudative” GN characterized by the presence of many intraglomerular neutrophils. Pyuria and even white cell casts may be observed in the urinary sediment.
 CLINICAL FEATURES
CLINICAL FEATURES
Clinical symptoms begin abruptly. Patients are usually afebrile with a latency period of 1 to 2 weeks following pharyngitis and 3 to 6 weeks after a skin infection. The most common presenting features in symptomatic patients are edema (85%) and gross hematuria (30% to 50%). Almost all the patients have microhematuria. The urine often has a unique smoky, cola or tea color. Hypertension is common (60% to 80%) but is usually mild to moderate; rarely, hypertensive encephalopathy and posterior reversible encephalopathy can develop even without a significant elevation in the serum creatinine concentration. The degree of renal failure is usually mild. Rapidly progressive GN is unusual, reported in less than 1% of children. Although many patients have significant proteinuria and a slightly depressed serum albumin level (at least in part due to intravascular volume expansion), fewer than 5% of symptomatic patients develop frank nephrotic syndrome. Severe complications, including pulmonary hemorrhage and cerebral vasculitis, have been reported. Spontaneous improvement typically begins within 1 week, with resolution of the edema occurring in 5 to 10 days and resolution of hypertension occurring in 2 to 3 weeks; however, the urinalysis may be abnormal for 1 year, rarely longer (Fig. 472-2 and eFig. 472.1  ).
).
 DIAGNOSTIC EVALUATION
DIAGNOSTIC EVALUATION
The serum C3 level is usually below 50% of normal value in patients with acute poststreptococcal glomerulonephritis (APSGN); this causes acute nephritis. However, up to 10% of APSGN patients have a normal C3 level. In patients with other forms of acute postinfectious GN, the percentage is even higher. Decreased serum C4 levels are unusual but have been reported, usually very early in the clinical course. A significant and persistent decrease in the C3 or C4 level should suggest an alternative diagnosis, such as systemic lupus erythematosus, bacterial endocarditis, shunt nephritis, or idiopathic membranoproliferative GN (Fig. 472-1). The serum C3 level typically returns to normal within 8 to 12 weeks after APSGN. Urinalysis generally shows hematuria, proteinuria, and cellular casts (both red and white cells), but rarely patients may have a normal urinalysis.
Confirmation of a recent streptococcal infection makes the diagnosis of APSGN most likely, with a few caveats. A positive throat culture is insufficient to confirm the diagnosis, as 20% of normal schoolchildren are carriers who will have positive throat cultures without disease. An antibody to the streptolysin O enzyme (ASO) is detected in 80% of children with antecedent pharyngitis, but the test is also positive in 16% to 18% of healthy children.  In the majority of patients, the clinical presentation and laboratory tests make the diagnosis of APSGN quite evident. There is no indication to perform a renal biopsy to confirm the diagnosis in such children. In approximately 10% of cases, acute postinfectious GN follows other antecedent diseases caused by a variety of infectious agents as summarized in eTable 472.1.
In the majority of patients, the clinical presentation and laboratory tests make the diagnosis of APSGN quite evident. There is no indication to perform a renal biopsy to confirm the diagnosis in such children. In approximately 10% of cases, acute postinfectious GN follows other antecedent diseases caused by a variety of infectious agents as summarized in eTable 472.1. 
 TREATMENT AND COMPLICATIONS
TREATMENT AND COMPLICATIONS
APSGN is an acute disease that resolves without specific medical therapy. Close monitoring and supportive care is essential to manage the acute nephritis syndrome until the glomerular injury spontaneously resolves. If not previously administered, antibiotics should be given to prevent the spread of the nephritogenic strain of Streptococcus to other individuals. In the acute phase, patients need to be hospitalized for observation and treatment if hypertension, edema, oliguria, elevated serum creatinine, electrolyte abnormalities, or other less common complications are present (Fig. 472-4). Fluid-overloaded patients often respond to loop diuretics. Fluids need to be balanced to prevent further intravascular volume overload, including appropriate sodium and fluid restriction. Urine output increases spontaneously within 5 to 10 days; severe oliguria beyond 2 weeks is extremely rare and is cause to question the diagnosis. Hypertension is typically not severe and can be managed with short-acting calcium channel blockers.
In patients with more severe renal insufficiency, hyperkalemia, hyperphosphatemia, and acidosis are likely to occur and will require medical management. These patients rarely need dialysis. Despite the old wives’ tale, bed rest is of no proven benefit once patients are well enough to ambulate. Corticosteroids and cytotoxic agents have no substantiated role, although there is limited anecdotal evidence of benefit for the rare patient with rapidly progressive glomerulonephritis associated with crescentic (in > 30% glomeruli) nephritis on biopsy.
 PROGNOSIS AND OUTCOMES
PROGNOSIS AND OUTCOMES
Spontaneous improvement should begin within 1 week, with the gross hematuria, oliguria, azotemia, and hypertension generally resolving within 4 weeks; low C3 within 2 months; proteinuria by 6 months; and microscopic hematuria by 1 year following diagnosis. Intermittent or orthostatic proteinuria may persist for up to 2 years (see eFig. 472.1  ). Prolonged hematuria up to 5 years has been observed but is rare. Traditionally, APSGN in children has been considered a completely reversible disease, and this is still generally true. In the exceptional patient with crescentic disease, prolonged oliguria, and massive proteinuria, recovery may not be complete.
). Prolonged hematuria up to 5 years has been observed but is rare. Traditionally, APSGN in children has been considered a completely reversible disease, and this is still generally true. In the exceptional patient with crescentic disease, prolonged oliguria, and massive proteinuria, recovery may not be complete.  Recently, cases of APSGN with superimposed histological lesions of thrombotic microangiopathy have been reported. Outcome may be poorer in this unique patient cohort, but insufficient data are available at this time. Recurrent APSGN is extremely rare but has been reported.7
Recently, cases of APSGN with superimposed histological lesions of thrombotic microangiopathy have been reported. Outcome may be poorer in this unique patient cohort, but insufficient data are available at this time. Recurrent APSGN is extremely rare but has been reported.7
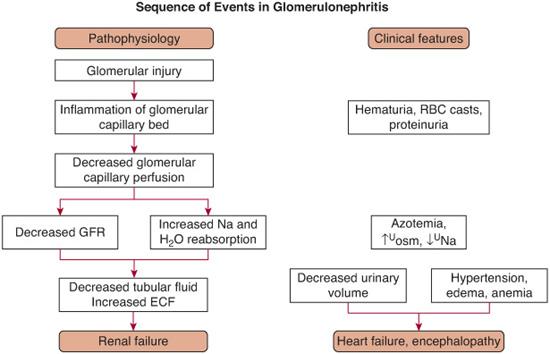
FIGURE 472-2. Sequence of pathophysiological events in a patient with acute glomerulonephritis and the associated clinical manifestations.
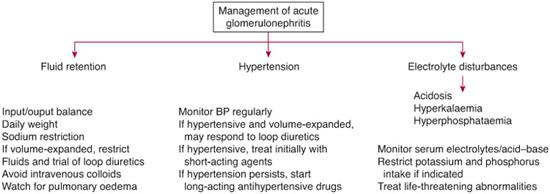
FIGURE 472-3. Management guidelines for the hospitalized child with acute glomerulonephritis. (From Smith JM, Faizan MK, Eddy AA. The child with acute nephritic syndrome. Postlethwaite RJ and Webb NJA (Eds). In: Clinical Paediatric Nephrology, 3rd Edition. New York: Oxford University Press, 2003, p. 374.)
GLOMERULONEPHRITIS WITH CHRONIC INFECTIONS
Immune responses may also induce glomerulonephritis (GN) in the face of certain chronic persistent infections. In pediatric patients, the most important examples of this are infective endocarditis, shunt nephritis, hepatitis B and C, and AIDS,8 as well as congenital infections with cytomegalovirus or toxoplasmosis that may present clinically as infantile nephrotic syndrome. In endemic tropical areas of the world, chronic infection with Plasmodium malariae, Schistosoma mansoni, and filariasis may cause more chronic forms of GN.
 INFECTIVE ENDOCARDITIS
INFECTIVE ENDOCARDITIS
Subacute bacterial endocarditis (usually caused by Streptococcus viridans) is less common with the decline in rheumatic fever, but acute bacterial endocarditis is now more common, especially among intravenous drug abusers. Staphylococcus aureus is the most common pathogen. Both serum C3 and C4 levels are depressed in 90% of patients. Histologically, the disease resembles APSGN, even in the small subset with neutrophil cytoplasmic antibodies. With appropriate antibiotic therapy, the glomerulopathy improves, but chronic renal failure may persist. Normalization of the serum complement levels is a good prognostic indicator.
 SHUNT NEPHRITIS
SHUNT NEPHRITIS
Shunt nephritis was first recognized in children with ventriculoatrial shunts used for hydro-cephalus. The risk of nephritis is reportedly 4% among patients with infected shunts. This complication is distinctly rare in patients with infected ventriculoperitoneal shunts. A similar form of nephritis occurs with infected vascular access grafts. Coagulase-negative Staphylococcus is the most common organism. Blood cultures may be negative, but the organism is often cultured from the graft itself. The renal disease frequently has an indolent presentation: gross hematuria is common, and 25% to 30% may develop nephrotic syndrome. Serum C3 and C4 levels are usually depressed (90%). Histologically, the lesion is a diffuse proliferative GN that resembles idiopathic MPGN. Treatment includes antibiotic therapy and removal of the infected shunt. The glomerular disease slowly resolves, but residual renal damage is common.
 CHRONIC HEPATITIS
CHRONIC HEPATITIS
An association exists between chronic hepatitis B (HBV) and membranous nephropathy. Less common renal manifestations associated with chronic HBV infection are membranoproliferative glomerulonephritis (MPGN), crescentic GN, and kidney disease related to polyarteritis nodosa. Introducing the hepatitis B vaccination during infancy has decreased the incidence of HBV-associated kidney disease.10 Children present clinically with nephrotic syndrome or with nonnephrotic proteinuria during the chronic carrier stage. Depressed serum C3 and C4 levels are not characteristic but may occur. In children, most renal complications remit spontaneously within 5 to 7 years, concomitant with the disappearance of HBe antigenemia and the appearance of HBe antibodies. Specific treatment guidelines for renal disease are not available, but antiviral drugs or interferon alpha is often recommended for persistent renal disease (see Chapter 308).11-13 Since these agents are nephrotoxic, renal function must be followed closely during therapy. Immunosuppressive therapy is contraindicated due to an increased risk of chronic active hepatitis, although corticosteroids have been used in a select subset of patients with severe vasculitis or crescentic GN.
Hepatitis C also can cause MPGN in adults, especially when it occurs in association with cryoglobulinemia. It has also been reported as a cause of MPGN in renal allografts and as a cause of membranous nephropathy and IgA nephropathy. Only isolated cases of hepatitis C–related renal disease have been reported in children.
 HIV-ASSOCIATED NEPHROPATHY
HIV-ASSOCIATED NEPHROPATHY
About 2% to 10% of patients infected with HIV have nephropathy. Most patients present with proteinuria, usually with nephrotic syndrome. This may be the first manifestation of HIV infection or may occur in patients with AIDS or AIDS-related complex. A unique variant of focal segmental glomerulosclerosis (FSGS) is found in 60% of patients, the vast majority of whom are of African descent with advanced HIV disease. In addition to the typical FSGS lesion, “collapsed” glomeruli (also seen in a small subset of patients with idiopathic FSGS), degenerated and micro-cystic tubules, interstitial edema and inflammation, and glomerular endothelial tubuloreticular inclusions (reminiscent of those associated with systemic lupus erythematosus [SLE] nephritis) are commonly observed features. Highly active anti-retroviral therapy (HAART) therapy appears to slow the rate of progression and may even prevent the development of nephropathy.14,15 Nephrotoxicity due to urinary crystal formation or tubular damage may occur with the use of certain antiretroviral drugs. In addition to the “collapsing” variant of FSGS, HIV-infected patients have an increased risk of developing other glomerulopathies, including IgA nephropathy, a glomerular microangiopathy that resembles hemolytic uremic syndrome; postinfectious glomerulonephritis (GN); membranoproliferative GN; and a lupuslike immune complex GN.
IGA NEPHROPATHY
IgA nephropathy is the most common primary cause of GN throughout the world. Although it may occur at any age, it is most common in the second and third decades of life and is rare in the first decade. However, it has been reported in children as young as 3 years of age. Even though the course of the disease is indolent in most patients, a significant number are at risk of ultimately developing end-stage renal disease (ESRD).
 EPIDEMIOLOGY
EPIDEMIOLOGY
The true incidence of IgA nephropathy is uncertain, as there are no reliable serological markers; a renal biopsy is the only way to establish the diagnosis. The disease appears to be more prevalent in Asians and Caucasians and is relatively rare in blacks, with the male-to-female ratio ranging from 2:1 to 6:1. Although IgA nephropathy is generally considered a sporadic disease, familial clustering has been observed in a pattern suggesting an autosomal dominant inheritance with incomplete penetrance. Specific genes have yet to be identified.
Most cases of IgA nephropathy are idiopathic, but multiple secondary causes are described, including severe liver disease (with regression following liver transplant); portosystemic shunts; enteropathies such as celiac disease and Crohn disease; chronic lung diseases, including obstructive bronchitis, cystic fibrosis, and idiopathic interstitial pneumonia; neoplasias such as small cell carcinomas and lymphoma; infections such as HIV, Mycoplasma, toxoplasmosis, and leprosy; dermatitis herpetiformis; and seronegative arthropathies. Lupus nephritis may also be characterized by significant IgA deposits. Many children with Henoch-Schönlein purpura have evidence of GN that histopatho-logically resembles IgA nephropathy. Some nephrologists consider these two entities to be different manifestations of the same disease. 
 PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
The pathophysiology and genetics of IgA nephropathy remain unclear. The glomerular IgA deposits are polymeric IgA1 derived from systemic rather than mucosal immune responses. IgA nephropathy has resolved when an IgA nephropathy kidney was transplanted into a patient with a different cause of ESRD, or following stem cell transplantation in a patient with IgA nephropathy. Compared to IgA from normal individuals, the IgA molecule itself is different, with a unique glycosylation pattern in the hinge region of the antibody characterized by reduced galactose residues. These modified IgA complexes may deposit in the mesangium, may interact with mesangial cells, and may activate C3 via the mannose-binding lectin pathway.
 CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
There are three classic clinical presentations of IgA nephropathy. Forty to 50% present with recurrent episodes of painless macroscopic hematuria. The onset of macrohematuria is often preceded 24 to 48 hours earlier by an upper respiratory tract infection; associations with gastroenteritis, sinusitis, and strenuous exercise have also been reported but are less common. The latency period between the onset of prodromal symptoms and macrohematuria is considerably shorter than that observed in patients with acute poststreptococcal glomerulonephritis (APSGN). The gross hematuria typically resolves within a few days, but microscopic hematuria and variable degrees of proteinuria typically persist. 
The first episode of macrohematuria may mimic APSGN. Recurrent episodes of macroscopic hematuria may occur in Alport syndrome during the first decade of life. Painless gross hematuria may be the initial presentation of children with MPGN. About 40% of patients present with an incidental finding of microhematuria and mild proteinuria (20% to 40%). A subset of this group will develop future episodes of gross hematuria, which occurs more frequently in children.  The interval between episodes is highly variable, ranging from a few months to many years. The frequency of the episodes decreases with age. Less than 10% present with nephrotic syndrome, an acute reversible nephritic episode, or rapidly progressive GN. Most of the acute nephritic episodes resolve without specific therapy, and the long-term prognosis remains relatively good. If a renal biopsy is performed during the acute phase, the degree of glomerular disease usually fails to explain the degree of renal dysfunction. Superimposed acute tubular necrosis is often evident.
The interval between episodes is highly variable, ranging from a few months to many years. The frequency of the episodes decreases with age. Less than 10% present with nephrotic syndrome, an acute reversible nephritic episode, or rapidly progressive GN. Most of the acute nephritic episodes resolve without specific therapy, and the long-term prognosis remains relatively good. If a renal biopsy is performed during the acute phase, the degree of glomerular disease usually fails to explain the degree of renal dysfunction. Superimposed acute tubular necrosis is often evident. 
 DIAGNOSTIC EVALUATION
DIAGNOSTIC EVALUATION
There are currently no reliable serological markers for IgA nephropathy. With the first episode of gross hematuria, serological studies are usually performed to rule out other causes of acute GN. Serum IgA levels may be elevated in 8% to 16% of children with IgA nephropathy; the percentage is higher in adults, but this test is not sufficiently specific to establish a diagnosis.  Most pediatric nephrologists would recommend a biopsy for IgA nephropathy patients who have impaired renal function, hypertension, or significant proteinuria (greater than 0.5 to 1.0 g/24 h), as these individuals are at increased risk of ultimately developing end-stage renal disease.
Most pediatric nephrologists would recommend a biopsy for IgA nephropathy patients who have impaired renal function, hypertension, or significant proteinuria (greater than 0.5 to 1.0 g/24 h), as these individuals are at increased risk of ultimately developing end-stage renal disease. 
 TREATMENT
TREATMENT
Optimal therapy for IgA nephropathy is controversial, but guidelines are beginning to emerge  (Fig. 472-2 and Fig. 472-4).16-20 The disease usually follows a benign course so that no therapeutic intervention is necessary. However, a subset of patients can develop progressive renal insufficiency. Risk factors include male sex, older age, elevated serum creatinine, hypertension, persistent proteinuria greater than 1.0 g/24 h, and the severity of the proliferative and sclerotic lesions on renal biopsy. Current management recommendations include the following:
(Fig. 472-2 and Fig. 472-4).16-20 The disease usually follows a benign course so that no therapeutic intervention is necessary. However, a subset of patients can develop progressive renal insufficiency. Risk factors include male sex, older age, elevated serum creatinine, hypertension, persistent proteinuria greater than 1.0 g/24 h, and the severity of the proliferative and sclerotic lesions on renal biopsy. Current management recommendations include the following:
1. Hypertensive patients should be treated with an ACE inhibitor (ACEi) plus/minus angiotensin receptor blockers (ARB), as these drugs control the blood pressure and may slow the rate of decline of glomerular filtration rate. Normotensive patients with proteinuria greater than 0.5 to 1.0 g/24 h may also benefit from such treatment with a decrease in proteinuria.
2. The benefit of treatment with omega-3 fatty acids in the form of fish oil (eicosapentaenoic acid and docosahexanoic acid) has not been firmly established due to conflicting clinical trial outcomes. It is thought to act as an anti-inflammatory agent. Compliance is poor due to fishy aftertaste.
3. Alternate-day steroids appear to benefit patients with slowly declining renal function. The optimal treatment duration is unclear; 6 months has been used in several clinical trials. Addition of cytotoxic agents appears to offer little benefit, although the efficacy of mycophenolate mofetil (MMF) is still under investigation.
4. Patients with aggressive crescentic GN are typically treated with corticosteroids and an immunosuppressive agent, as the prognosis is otherwise poor. 
5. Tonsillectomy has been proposed as a therapeutic option but there is a lack of evidence of long-term benefit when performed in patients without evidence of tonsil-associated clinical symptoms. 6) Patients with isolated microscopic hematuria and urinary protein-to-creatinine ratios less than 0.6–0.8 mg/mg (males and females respectively) require no treatment except monitoring.
 PROGNOSIS
PROGNOSIS
IgA nephropathy is a disease with a highly variable outcome. Complete remission occurs in less than 10% of patients. For patients with self-limited episodes of recurrent gross hematuria who do not develop significant proteinuria, the long-term outcome is quite good. Risk factors for progressive disease include persistent and significant degrees of proteinuria, hypertension, and an elevated serum creatinine.21 In the high-risk patient group, renal function slowly deteriorates. After 20 to 25 years of follow-up, 20% to 30% develop end-stage kidney disease while 20% have reduced renal function. Following kidney transplantation, IgA deposits commonly recur in the allograft (20% to 75%), but recurrent disease is not a common cause of graft failure.
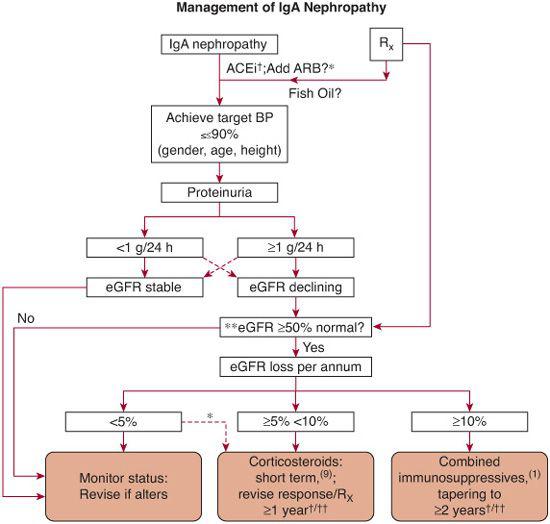
FIGURE 472-4. An algorithm of recommended treatment options for IgA Nephropathy. †therapy with efficacy of evidence base grade 1 data; *therapy with high a priori evidence to use but not tested independently in randomized, controlled trials; ††remission in erythrocyturia and proteinuria, follow no <6 and 12 mo, respectively, after starting immunosuppressive drugs1; **there is no evidence that immunosuppressive drugs can benefit declining function in patients starting therapy with >50% loss in GFR.1 Broken lines denote less frequently encountered scenarios. eGFR, estimated (calculated) glomerular filtration rate; ACEi, angiotensin converting enzyme inhibitor; ARB, angiotensin receptor blocker. (From Ballardie, F. W. Quantitative appraisal of treatment options for IgA nephropathy. J Am Soc Nephrol 2007;18: 2806-9.)
MEMBRANOPROLIFERATIVE GLOMERULONEPHRITIS (MPGN)
MPGN, less commonly called mesangiocapillary GN, is not a distinct disease. It is a specific form of glomerulonephritis (GN) characterized morphologically by (1) thickening of the glomerular basement membrane (GBM) caused by immune complex deposition or interposition of mesangial cell cytoplasm in the GBM, and (2) by hypercellularity caused by proliferation of mesangial cells and the influx of leukocytes, which produces a typical lobular appearance of the glomerular tuft. MPGN is frequently idiopathic in children but may be secondary to a variety of other entities, as shown in Figure 472-5.
The idiopathic varieties are classified as two distinct and unrelated diseases. Type I is an immune complex disease characterized by immune deposits containing C3 and immunoglobulins (IgG > IgM) deposited along the subendothelial space of the GBM. Type III appears to be a histological variant of type I, with codeposition of prominent subepithelial deposits. In general, types I and III are quite similar and are considered together here. In contrast, type II MPGN, also known as dense intramembranous deposit disease, is clearly a distinct entity characterized ultrastructurally by the presence of dense ribbonlike deposits within the GBM. The nature of this dense material remains unknown, but it appears to activate the alternative complement cascade, with C3 characteristically lining the outer aspect of these deposits. Immunoglobulins are typically absent.
 EPIDEMIOLOGY
EPIDEMIOLOGY
Idiopathic membranoproliferative glomerulonephritis (MPGN) usually presents between the ages of 8 and 30 years; it is rare under the age of 5 years (although cases as young as 15 months have been reported) and mainly affects Caucasians. Although uncommon, familial clusters are reported. The incidence of idiopathic MPGN, particularly type II, appears to have decreased over the past decade. In adults, this may be explained by the recognition that many patients previously classified as idiopathic MPGN actually are chronically infected with hepatitis C. The reason for the declining pediatric incidence of idiopathic MPGN is unclear.
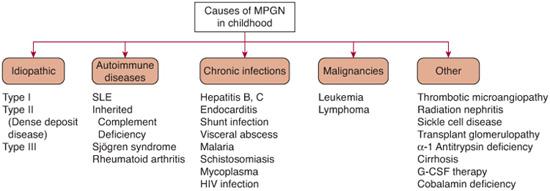
FIGURE 472-5. Causes of membranoproliferative glomerulonephritis (MPGN) in the pediatric population. SLE, systemic lupus erythematosus.
 PATHOPHYSIOLOGY
PATHOPHYSIOLOGY
The pathophysiology of idiopathic MPGN remains enigmatic. Type I/III is considered an immune complex–mediated glomerular disease. A small subset of patients who have inherited complement deficiencies are predisposed to develop MPGN, especially those with factor H deficiency.  C3 nephritic factor (C3NeF) is an autoantibody that reacts with and stabilizes the convertase activity of C3bBb, leading to ongoing C3 activation. C3NeF may be present in all types of MPGN but is most common in type II. Whether it plays a pathogenic role, is a disease marker, or is a secondary epiphenomenon remains unclear.
C3 nephritic factor (C3NeF) is an autoantibody that reacts with and stabilizes the convertase activity of C3bBb, leading to ongoing C3 activation. C3NeF may be present in all types of MPGN but is most common in type II. Whether it plays a pathogenic role, is a disease marker, or is a secondary epiphenomenon remains unclear.
 CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
Twenty to 30% of children with MPGN present with an acute nephritic syndrome that initially mimics acute poststreptococcal glomerulonephritis (APSGN). The presence of nephrotic syndrome and a depressed serum C4 level are early clues that differentiate MPGN from APSGN. Another 20% to 40% of children are diagnosed following an incidental finding of proteinuria and hematuria. Nephrotic syndrome is a presenting feature in 30% to 50% of children, and even more children will develop the syndrome during the course of the disease. Subtle, but not substantial, differences exist between the clinical features at presentation of types I and II. Some patients with MPGN II have partial lipodystrophy (Barraquer-Simons disease).
 DIAGNOSTIC EVALUATION
DIAGNOSTIC EVALUATION
A renal biopsy is required to establish the diagnosis of membranoproliferative glomerulonephritis (MPGN), although a characteristic clinical presentation with persistent hypocomplementemia is highly suggestive. Type I patients have low serum C3 levels and borderline or low C4 levels. The complement profile in type II patients suggests alternative pathway activation with low C3 and normal C4 levels. Overall, the serum C3 level is decreased in 75% of all patients at the time of initial presentation. The levels are variable over time and do not have prognostic significance. Normalized C3 levels 6 to 8 weeks after the onset of acute nephritis will help differentiate APSGN from MPGN (Fig. 472-1).
 TREATMENT AND COMPLICATIONS
TREATMENT AND COMPLICATIONS
Optimal therapy remains uncertain. Randomized clinical trials have shown no benefit of any treatment, but the duration of trials has been relatively short; this is significant because MPGN is typically a chronic, slowly progressive disease process, so the benefit of treatment may not be evident unless given for years.22 For many children without nephrotic syndrome, renal function may remain stable for many years without specific therapy. The consensus in North America is that children with idiopathic MPGN type I and significant proteinuria will do better if treated with alternate-day steroids for 3 to 10 years. A variety of different protocols have been tried, but most involve a dose of 2 mg/kg every other day (maximum 60 mg) for at least 1 year, with various tapering schedules thereafter. Hypertension is a frequent complication that requires treatment. The role of antiplatelet therapy with aspirin or dipyridamole remains unclear and is generally not recommended. Limited and conflicting data on the use of cytotoxic drugs preclude recommendation.
There are few data to support the use of immunosuppression for dense deposit disease.16,23 ACEi and angiotensin receptor blockers (ARB) are recommended for proteinuria and hypertension control. Plasma exchange therapy may have a role in the patient subgroups with inherited complement factor H and deficiency and possibly those with significant renal disease associated with C3NeF.
 PROGNOSIS AND OUTCOMES
PROGNOSIS AND OUTCOMES
Limited data are available on the long-term outcome of children with idiopathic MPGN. For many patients, the disease carries a poor prognosis, particularly if associated with nephrotic syndrome. Overall, 50% to 60% of untreated patients develop ESRD within 10 to 15 years; less than 10% undergo spontaneous remission. 
RAPIDLY PROGRESSIVE (CRESCENTIC) GLOMERULONEPHRITIS (RPGN)
RPGN is a clinical syndrome characterized by sudden onset and rapid decline in renal function.24 Renal histology shows extensive crescent formation, typically in more than 50% of the glomeruli. Without specific therapy, most patients progress to ESRD within a period of weeks to months. Early treatment offers the best chance for renal recovery, so in suspected cases, early diagnosis is a nephrologic emergency.
 EPIDEMIOLOGY
EPIDEMIOLOGY
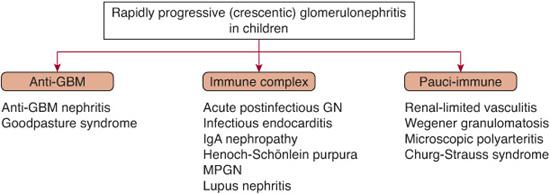
RPGN is a rare disorder in childhood that is classified into three groups based upon the immunopathologic features (Fig. 472-6): (1) anti-GBM nephritis (12%), (2) immune complex nephritis (45%), and (3) pauci-immune disease associated with ANCA (42%). Less commonly, other forms of proliferative glomerulonephritis may present as severe crescentic disease.
 CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
Children with RPGN either present with gross hematuria (50% to 85%), edema (13% to 80%), anemia (70%), and hypertension (63% to 85%), or in a more insidious manner, they may present with symptoms due to chronic renal failure. The urinalysis typically reveals an active urine sediment with hematuria, proteinuria, and cellular casts. Depending on the etiology, extrarenal manifestations may also be present. Pulmonary hemorrhage suggests a diagnosis of anti-GBM nephritis or Wegener’s granulomatosis and may be fatal if treatment is delayed.
 DIAGNOSIS
DIAGNOSIS
Serological studies can be very helpful in establishing a tentative diagnosis, but biopsy confirmation is mandatory. Initial tests should include a C3, C4, antinuclear factor (ANA), anti-GBM antibody, and ANCA antibodies. Biopsy establishes the diagnosis in most patients and provides valuable information about the relative degree of acute (potentially treatable) and chronic histological changes (Fig. 472-6). If the kidney is damaged beyond the point of repair, immunosuppressive therapy is futile, at least for the renal component of the disease.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


