Global Burden of Respiratory Infections and Mucosal Immunology of the Respiratory Tract
Jay K. Kolls and Joseph P. Mizgerd
THE BURDEN OF LUNG INFECTIONS IN THE WORLD
The World Health Organization (WHO) has been tracking the global burden of infectious diseases for several decades. During that time, mortality resulting from acute respiratory infections has been increasing, surpassing diarrheal disease in the late 1990s to become the number one killer of children worldwide. Since 1990, the WHO has used the Burden of Disease Project to assess disease-related morbidity and mortality, including the summary measure of disability-adjusted life years (DALYs) lost, which takes into account the degree and duration of morbidity in addition to mortality. When data are assessed by the DALYs measure, lung infections are again remarkably prominent. The attributable morbidity and mortality of acute respiratory infections exceeds HIV/AIDS, cancer, and heart disease. Of particular note is that these data do not include morbidity and mortality resulting from tuberculosis or to AIDS-related pneumonias, thereby underestimating the true impact of respiratory infections (Fig. 504-1). When stratified by age, there is a large and disproportionate burden of morbidity and mortality attributable to lower respiratory tract infections (LRIs) in the 0-to-4 and 5-to-14 age groups. Across the decades, even for the youngest patients, the burden of disease from LRIs exceeds that from malaria or diarrheal disease (Fig. 504-2). Although there is a strong association of LRIs with poverty, LRIs account for remarkable morbidity and mortality in wealthy countries as well.1 In the wealthiest countries, LRIs cause a greater burden than any other infectious disease. Although effective vaccines are available for specific pathogens such as Hemophilus influenza b and some serotypes of Streptococcus pneumoniae, the number of organisms capable of causing lung infections is too numerous to feasibly vaccinate against each. An approach that bolsters lung immunity per se may have the greatest impact in reducing morbidity and mortality against LRIs. It is imperative to advance our understanding of both innate and adaptive immunity to pathogens in the lung.
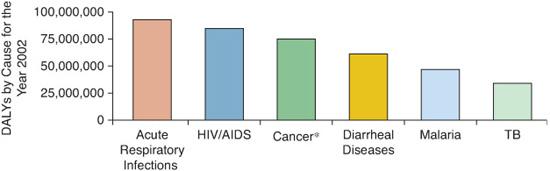
FIGURE 504-1. Global burden of disease as measured by the disability-adjusted life years (DALYs). Acute respiratory infections cause more morbidity than HIV/AIDS, cancer, diarrhoeal disease, malaria, or tuberculosis (TB).
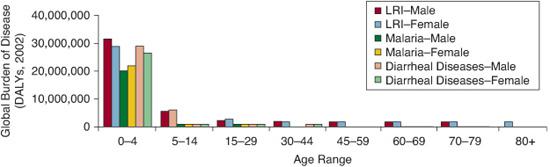
FIGURE 504-2. Disproportionate burden of disease in children ages 0 to 4 and 5 to 14. Acute respiratory infection causes the greatest combined morbidity and mortality in children.
MUCOSAL IMMUNOLOGY OF THE RESPIRATORY TRACT
With the exceptions of neonatal pneumonias in which the lungs become seeded by hematogenous dissemination of group B streptococcus or Escherichia coli, most pneumonias result from aspiration of organisms colonizing the upper airways.2 If this inoculum is small or of lower virulence, innate immunity consisting of cough, mucociliary clearance, antimicrobial peptides, and/or resident airway or alveolar macrophages can eradicate the infection. However, if the inoculum is large (as can occur with oral anerobes in children with poor dentition) or if more virulent pathogens (such as encapsulated bacteria) are aspirated, then the end result can be pneumonia. The host inflammatory response, or recruitment of innate immunity reserves, is critical to the development and outcome of clinical pneumonia. For example, inflammation mediated by early cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) can result in fever and malaise, but these cytokines are also essential for host defenses against some pathogens in the lungs.2 Local production of granulocyte-colony stimulating factor (G-CSF) is essential to eradicate many pathogens,3,4 in part by regulating granulopoiesis and neutro-phil apoptosis. In fact, the local G-CSF response may explain why many patients with bacterial pneumonia present with elevated white blood counts in peripheral blood.3 These inflammatory responses can also regulate the severity of illness by altering ventilation-perfusion relationships leading to hypoxemia as well as regulating lung compliance, in part by regulating the amount of exudate in the alveolar spaces in the lung.
NEUTROPHILS AND PNEUMONIA
Normally, neutrophils constitute less than 1% of the cells in the alveolar air spaces. However there is a large number of marginated neutrophils in the extensive network of pulmonary capillaries, and these can be recruited to the alveolar air space very rapidly. Although some strains of Staphylococcus aureus are cleared by macrophages in the absence of recruited neutrophils, neutrophil emigration into the lung is critical for most bacteria.2 In addition, neutrophils have been shown to be essential for control of viral and fungal pneumonias.5,6 Neutrophils kill extracellular pathogens through their ability to phagocytize organisms and to generate reactive oxygen species (ROS). Neutrophils also express antimicrobial proteins in their granules including defensins, which have direct microbicidal activity,7 and lipocalin-2, which binds certain bacterial siderophores and chelates iron necessary for bacterial growth.8 In addition to phagocytosis, neutrophils can form neutrophil extracellular traps (NETs), which consist of DNA and proteins that can immobilize and kill bacteria.9
INITIATION OF AIRWAY INFLAMMATION IN PNEUMONIA
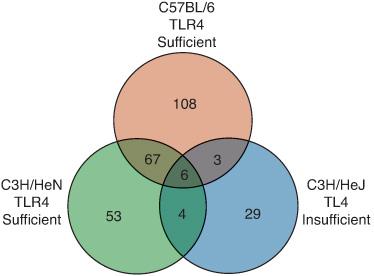
There has been an explosion in knowledge over the last decade of how the host recognizes invading pathogens. It was already known by the mid-1990s that lipopolysaccharide or endotoxin, a major component of the gram-negative cell wall, was critical in eliciting host immunity to gram-negative pathogens. In fact, mice with a naturally occurring mutation in the lps gene locus were highly attenuated for the inflammatory response to purified LPS, as measured by the ability to produce TNF-α or IL-1β. However, these mice were also highly susceptible to gram-negative bacterial pneumonia, demonstrating the critical nature of this innate recognition pathway.10 Using positional cloning, Poltarek and colleagues showed that the product of the lps gene was a member of the Toll-like receptor family of proteins, TLR4, which is required for the recognition of LPS.11 Whereas wild-type mice induce over 200 genes in the lung four hours after instillation of gram-negative bacteria, mice with a mutation in tlr4 only induce 20 to 30 genes (Fig. 504-3).10 Thus, signaling through this single receptor accounts for well over 70% of the gene expression during experimentally induced gram-negative bacterial pneumonia. Humans with mutations in Tlr4 show reduced airways inflammation due to inhaled endotoxin,12 increased mortality due to sepsis,13 and increased risks of severe respiratory syncytial virus infection.14
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


