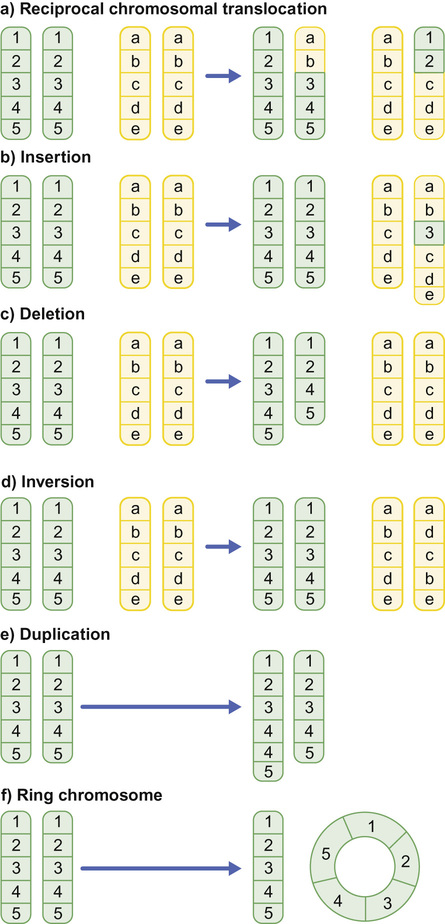
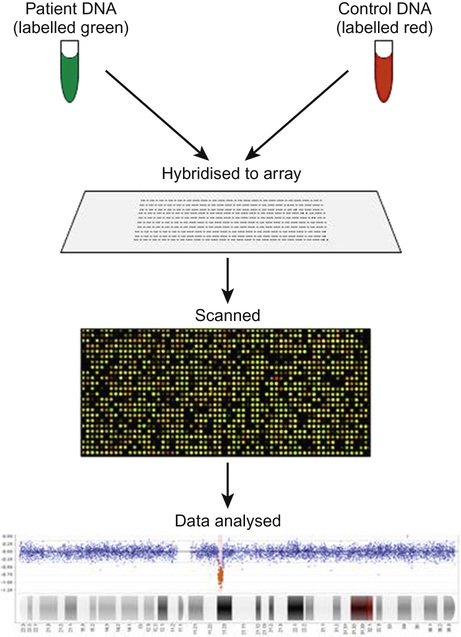
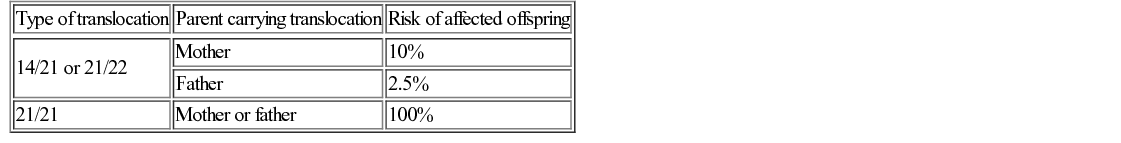
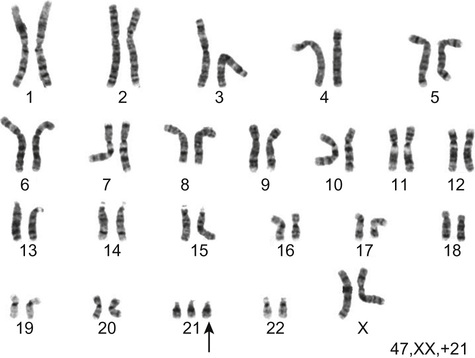
Richard H Scott, Shereen Tadros The term ‘chromosome’ comes from the Greek for ‘colour’ and ‘body,’ and was coined in the late 19th century in reference to their staining behaviour when dyes were applied. Chromosomes are string-like bodies present in the nucleus of every nucleated cell. Each chromosome consists of DNA that is tightly coiled around proteins known as histones that support its structure. Much of what we know about chromosomes comes from observations during cell division, when the DNA becomes more tightly coiled and is thus visible under a microscope. Humans normally have 22 pairs of numbered chromosomes – autosomes – and one pair of sex chromosomes – XX or XY. Each parent contributes one chromosome to each pair; therefore, children get half of their chromosomes from their mother and half from their father. Each chromosome has a constricted region known as a centromere. The regions on either side of the centromere are the chromosome’s arms. The shorter arm is the ‘p arm’ (for ‘petit’ or small) and the longer arm is the ‘q arm’ (as next letter in the alphabet!). The ends of the chromosomes are called telomeres. Each chromosome arm is divided into regions or bands which can be seen microscopically with special stains. The bands are labelled counting out from the centromere, with p1 and q1 being closest to the centromere. Within the bands, there are sub-bands (only seen at very high resolutions), which are also numbered outwards from the centromere. Chromosome abnormalities can be classified into two groups. Numerical abnormalities involve a missing (monosomy) or extra (trisomy or tetrasomy) whole chromosome from a pair. Examples include Turner’s syndrome (monosomy X) and Down’s syndrome (trisomy 21) (Fig. 9.1). Structural abnormalities can take several forms (Fig. 9.2): • Translocation – a portion of one chromosome is transferred to another chromosome • Insertion – a segment of a chromosome is inserted into another position on a chromosome • Deletion – a portion of the chromosome is missing, e.g. 22q11 deletion • Duplication – a portion of the chromosome is duplicated, meaning there is extra genetic material • Ring – a chromosome has formed a circle. This can happen with or without a loss of genetic material. As chromosomes are only visible microscopically in dividing cells, chromosome analysis involves taking some non-dividing cells (usually peripheral blood lymphocytes) and culturing them to encourage cell division. The cytogeneticist then stains and subsequently examines the cells for any extra or missing chromosomes, or large scale structural chromosomal abnormalities. This method will miss any changes smaller than 5–10 megabases (5–10 million bases) in size, so is unsuitable for the detection of most microdeletions or duplications. It may be helpful in identifying the structural basis for abnormalities detected by other methods, including array comparative genomic hybridization. Array CGH, or microarray (Fig. 9.3), is a relatively new technique which looks for chromosomal copy number variations (CNVs) by comparing a patient’s DNA to normal control DNA. It has taken over from karyotype as the first line test for most chromosome abnormalities in paediatric practice. In CGH, the patient DNA is labelled green and the normal control DNA is labelled red. Thousands of different probes (fragments of DNA) which bind specifically to regions spanning the whole genome are immobilized on a slide, the array. The array is immersed in a solution containing equal proportions of the fluorescently-labelled test and control DNA allowing green ‘patient’ DNA to compete with red ‘control’ DNA to hybridize with each of the probes. At probes where there is a deletion (i.e. lower patient copy number than control), the array appears red, and where there is duplication, the array appears green. The array is scanned with a high resolution camera and the data interpreted using computer software. Array CGH is typically able to detect deletions and duplications larger than approximately 50,000 bases. This is a resolution more than 100 times higher than karyotyping and allows the detection of microdeletions or duplications as well as other larger chromosome copy number abnormalities, including aneuploidies. However, array CGH cannot detect balanced chromosomal rearrangements and does not distinguish unbalanced chromosome rearrangements caused by different mechanisms; for example, it does not distinguish conventional trisomy 21 from trisomy 21 caused by Robertsonian translocation. Fluorescence in situ hybridization (FISH) is a modification of conventional chromosome analysis using fluorescently labelled probes. It allows targeted testing for copy number variations and structural rearrangements that would otherwise be beyond the technique. Chromosomes are immobilized and denatured on a microscope slide and exposed to a solution containing a fluorescently labelled probe specific to a specific chromosomal region. After hybridization (the formation of a double strand of DNA from complementary single strands), the slide is washed and examined microscopically. Where the probe has hybridized, fluorescent spots are seen over the relevant chromosome. For example, if a child were suspected of having 22q11 deletion syndrome, FISH using a 22q11-specific probe would show only one pair of fluorescent spots, rather than two. As FISH testing only detects abnormalities in the region targeted by the chosen probe, it requires clinical recognition of the likely causative mechanism. For this reason, it has largely been replaced by array-CGH testing when detecting microdeletions. It is now most commonly used for rapid testing for aneuploidy (chromosome number not a multiple of the haploid number, i.e. of 23 chromosomes in humans) and for the follow-up of array-detected abnormalities. This is another technique commonly used for rapid aneuploidy testing and for follow-up of array-detected abnormalities. Mitosis is the process by which most cells replicate and results in the production of two identical daughter cells, each with a diploid genome (46 chromosomes). Meiosis is the process by which gametes are formed. It involves two successive cell divisions and results in cells with a haploid genome (23 chromosomes). However, errors can and do arise in this process. Numerical chromosome abnormalities generally happen because of non-disjunction during meiosis. This is when either the two paired chromosomes (in meiosis I) or the two sister chromatids (in meiosis II) fail to separate and segregate into separate cells. The resultant gametes therefore have either too many (24) or too few (22) chromosomes. Following fertilization, the zygote is either trisomic or monosomic for the relevant chromosome. Embryos can be trisomic for every chromosome and most will spontaneously abort. The only commonly survivable trisomies are discussed below. Rarely, there are mosaic aneuploidies affecting other chromosomes (see below). Non-disjunction can happen in both men and women. However, most cases of trisomy seem to take place because of non-disjunction at meiosis I in the mother. It is presumed that this might be due to the fact that this stage is very long in women – starting before birth and ending at ovulation. The risk of Down’s syndrome (trisomy 21) increases with maternal age; from 1 in 1500 for a 20-year-old woman, to 1 in 30 for a 45-year-old woman. There is a similar age-dependence with all other aneuploidies except for Turner’s syndrome. Turner’s syndrome (45, X) occurs by a different mechanism – anaphase lag, whereby one of the sex chromosomes (X or Y) moves too slowly to the pole of a daughter cell during cell division. It therefore ends up outside the nucleus and is broken down. It can happen during meiosis when gametes are being made, or during early mitotic divisions of the embryo. Chromosome mosaicism refers to the presence of two cell lines with differing numbers of chromosomes in each, usually one is abnormal and one normal. Examples include mosaic Down’s syndrome (e.g. 46, XX/47, XX+21) and mosaic Turner’s syndrome (45, X/46, XX). Mosaicism usually results in milder phenotypes. Mosaicism can occur when the abnormal cell line arises post-zygotically or when aneuploidy is present from conception but some cells revert to a normal karyotype, for example by losing one copy of a trisomic chromosome – so-called trisomy rescue. A translocation is described as ‘balanced’ if there is no net loss or gain of genetic material or ‘unbalanced’ if there is deletion or duplication of genetic material. Balanced translocations are important to recognize as they can result in the transmission of an unbalanced chromosome translocation to offspring. Occasionally, balanced chromosome translocations can cause disease themselves, for example if a translocation breakpoint disrupts a disease gene. These arise when any two chromosomes swap non-homologous segments. A carrier of a reciprocal translocation may potentially have offspring with trisomy of one of the translocated segments and monosomy of the other. These are translocations involving the ‘acrocentric’ chromosomes: 13, 14, 15, 21, 22 and Y. These chromosomes have their centromeres close to one end. In a Robertsonian translocation, inappropriate non-homologous recombination means that two acrocentric chromosomes join to form a single fusion chromosome with breakpoints on the short arm, just above the centromere. As with reciprocal translocations, balanced Robertsonian translocations are important to recognize because they can cause transmission of an unbalanced chromosome complement to offspring. In the case of Robertsonian translocations, this causes trisomy or monosomy for the affected chromosome. The most frequently encountered Robertsonian translocations involve chromosome 21 and can predispose to trisomy 21 in offspring, as discussed below. Most autosomal aneuploidies are fatal in utero. Three are survivable to term: Trisomy 13, 18 and 21. Children with trisomy 21 (Down’s syndrome) are believed to have the most favourable outcome, as chromosome 21 has fewer genes than the other autosomes (it is short and has a low gene density). Some 50% of babies die in the first month and most of the rest in the first year. Polydactyly and cardiac abnormalities are common, along with midline abnormalities of the head and face: Most babies die in the first year of life. Those that survive make little developmental progress. Dysmorphic features can be subtle and include ear anomalies, clinodactyly, overlapping fingers, micrognathia and rocker-bottom feet. Growth is poor, both antenatally and postnatally, and the head is small. Cardiac and renal abnormalities are common. Answer 9.2 B. Karyotype in the child with Down’s syndrome More than 95% of all Down’s syndrome is caused by non-disjunction resulting in simple trisomy 21. This has a low risk of recurrence in future pregnancies (~1% or the mother’s age-related risk, whichever is higher). Approximately 2% of Down’s syndrome is mosaic. The phenotype in these cases is often milder. The recurrence risk is low. Approximately 2% of Down’s syndrome is caused by unbalanced Robertsonian chromosome translocation. In these cases, one parent may carry a balanced translocation, in which case the risk of having a child with trisomy 21 is much higher. The risks seen with the common translocations involving chromosome 21 are shown in Table 9.1. To determine the recurrence risk in a child with aneuploidy, it is important to differentiate conventional trisomy 21 (which has occurred as a result of non-disjunction) from Down’s syndrome caused by an unbalanced Robertsonian translocation, which can result from the unbalanced transmission from a parent with a balanced translocation. This can be achieved by karyotyping or FISH analysis, which directly visualize the chromosomes. Parental microarray will not detect a balanced translocation. While aneuploidies are associated with increased maternal age, ovarian function testing will not be helpful. Skin biopsy for chromosomal mosaicism is also unlikely to be useful. The clinical features will not assist in determining the future risk. This is the only autosomal trisomy frequently associated with survival into adulthood. Children are often hypotonic at birth. Dysmorphic features that allow clinical recognition include: epicanthic folds, upslanted palpebral fissures, protruding tongue, sandal gap (between the great and second toes), Brushfield’s spots in the iris, single palmar crease. Cardiac and gastrointestinal abnormalities are common, and include atrioventricular septal defect (AVSD), ventricular septal defect (VSD), duodenal atresia and Hirschsprung’s disease. Affected children have mild to moderate learning difficulties. Behaviour is often problematic when older (see Chapter 24, Emotions and behaviour). Long-term problems include short stature, obesity, increased risk of leukaemia and solid tumours and Alzheimer’s disease. The Y chromosome determines maleness – a person is male if he has a Y chromosome, regardless of how many X chromosomes are present. This is the only survivable monosomy. Nonetheless, most fetuses abort spontaneously and can be grossly hydropic. The clinical features are described in Chapter 12, Growth and puberty. XXY is the commonest sex aneuploidy in humans – the prevalence is around 1 in 500 males. Boys with this condition have a normal facial appearance and may not be diagnosed until later in life. Babies and young children may present with hypospadias, micropenis, cryptorchidism or developmental delay. School-aged children may have language delay, learning difficulties and behaviour problems. Older children may present with incomplete pubertal development, gynaecomastia or small testes. Adults present with infertility. Testosterone replacement therapy starting at around 12 years of age can improve behaviour and learning, as well as the development of secondary sexual characteristics. Most men with Klinefelter’s syndrome do not produce sperm. However, intra-cytoplasmic sperm injection (ICSI) can help with fertility in men who produce minimal sperm. The phenotypic effect of a deletion or duplication depends on the genes it encompasses. In general, deletion is more likely to have a phenotypic effect than duplication and there are more chromosome deletion than duplication syndromes recognized. As described above, with conventional karyotype many of the common pathogenic deletions and duplications are too small to be visible on conventional karyotyping. Historically, they required the clinician to recognize the disorder and request the correct FISH test. As array CGH has a much higher resolution than karyotype and tests across the whole genome for microdeletions and duplications in a single test, it has replaced karyotype as the first line test for suspected chromosomal abnormalities in paediatrics. With the advent of microarray testing, an increasing number of recurrent microdeletion and microduplication syndromes are being recognized. The best delineated syndromes, however, remain those that are clinically recognizable and detectable using karyotype and FISH. DiGeorge or velocardiofacial syndrome is caused by deletion at the 22q11 region of one copy of chromosome 22. The deletion follows autosomal dominant inheritance. It is helpful to test the parents as they may have a milder phenotype and not be aware of it. However, the majority of cases are de novo. Patients may have a long face, narrow palpebral fissures and over-folded ear helices. However, the facial phenotype is often subtle. Affected individuals can have very variable medical complications affecting almost every system:
Genetics
Chromosomal disorders
Chromosomes
Types of chromosomal abnormalities
Chromosome testing
Karyotyping
Array comparative genomic hybridization (aCGH)
Fluorescence in situ hybridization
Quantitative fluorescent PCR (QF-PCR)
How do chromosomal abnormalities happen?
Non-disjunction
Anaphase lag
Mosaicism
Chromosome translocations
Reciprocal chromosome translocations
Robertsonian translocations
Autosomal aneuploidies
Trisomy 13 (Patau syndrome)
Trisomy 18 (Edwards syndrome)
Trisomy 21 (Down’s syndrome)
Sex chromosome aneuploidies
45, X (Turner’s syndrome)
47, XXY (Klinefelter’s syndrome)
Partial chromosomal deletions and duplications
22q11 deletion syndrome (DiGeorge or velocardiofacial syndrome)
Genetics
Chapter 9
Learning objectives
After reading this chapter the reader should: