Genetics of Heart Disease
Kathleen M. Ruppel
Most congenital heart defects (CHDs) are due to the interaction of genetic predisposition with environmental influences. Historically, evidence for a genetic contribution to CHD came from studies of familial recurrence and the association of certain types of CHD with specific chromosomal abnormalities, whereas epidemiologic data supported a role for environmental factors as well.1 More recently, our understanding of the genetic contribution to CHD has advanced significantly, and it is clear that genetics plays a more important role than previously understood. Molecular genetic analysis of families with documented Mendelian transmission of CHD has led to the identification of disease-causing mutations in several cardiac transcription factors and signaling molecules. Studies of cardiac development in model organisms have led to the discovery of a number of genes required for heart formation. Searching for mutations in these “candidate” genes in patients with familial or isolated forms of CHD has yielded insight into the genetic basis of certain defects.
Clinical testing is increasingly available for many of the genetic alterations that underlie CHD, and information derived from these tests is affecting management of CHD patients and their families. Determining whether there is an underlying genetic defect in the patient with CHD is important for several reasons. Twenty to 30% of infants with CHD will have at least one other developmental anomaly.2,3 Diagnosis of a specific genetic syndrome facilitates early identification and treatment of other organ system involvement. Defining causative mutations may also provide prognostic information in defects for which there are genotype-phenotype correlations. In addition, identifying specific genetic defects allows for screening of family members who may be at risk for CHD, and also provides crucial information on reproductive risks for the family of the patient with CHD. As treatment of CHD improves and more CHD patients survive into adulthood, genetic diagnoses will aid these patients in their own reproductive decisions. For updated information on genetic testing availability, the reader is referred to online resources such as GeneTests (www.genetests.org).
This section contains a review of what is currently known about the contribution of genetics to both syndromic and isolated cardiovascular malformations. The genetic basis of cardiomyopathies and arrhythmias is discussed in other sections. For more extensive clinical descriptions of the syndromes discussed in this chapter, the reader is referred to text4 and online resources (see the Online Mendelian Inheritance in Man [OMIM] Web site at www.ncbi.nlm.nih.gov/omim/).
CONGENITAL HEART DEFECT ASSOCIATED WITH GENETIC SYNDROMES
CONGENITAL HEART DEFECT ASSOCIATED WITH CHROMOSOMAL ABNORMALITIES
In population-based studies, approximately 13% of infants5 and up to 36% of stillborn and spontaneously aborted fetuses5,6 with cardiac defects had chromosomal abnormalities. Conversely, at least 30% of children with chromosomal anomalies have some form of congenital heart defect (CHD).7 Thus, when evaluating the patient with known or suspected chromosomal disorders, it is important to look for CHD, just as it is crucial to consider cytogenetic testing for certain CHD patients, especially those with other organ system involvement.
 ANEUPLOIDY SYNDROMES
ANEUPLOIDY SYNDROMES
Changes in chromosome number (aneuploidy) were among the first identified genetic causes of congenital heart defect (CHD), and can involve either autosomes or sex chromosomes. Several of the more common disorders are discussed in this section and listed in Table 481-1.
Trisomy 21 (Down Syndrome)
Trisomy 21 is the most common autosomal chromosomal abnormality affecting live-born infants (1/800; see Chapter 174). The prevalence of congenital heart defect (CHD) in this population is approximately 40% to 50%. Complete atrioventricular septal defect (AVSD; also termed endocardial cushion defect, atrioventricular canal defect) accounts for 40% of all CHD in Down syndrome patients, and this figure increases to almost 60% when partial AVSD (primum atrial septal defect, inlet or canal-type ventricular septal defect) is included.8 Other associated lesions are shown in Table 481-1. The American Academy of Pediatrics recommends routine echocardiographic screening of neonates with Down syndrome to facilitate early diagnosis of CHD and intervention9 because of increased risk of pulmonary vascular disease in this population.10,11
The mechanism by which an extra copy of chromosome 21 results in the pathogenesis of CHD in Down syndrome is unknown. 
Although 70% of patients with complete AVSD have Down syndrome, analysis of kindreds with autosomal-dominant AVSD has excluded linkage to chromosome 21,16,17 suggesting that distinct genetic etiologies account for this cardiac defect in Down syndrome versus nonsyndromic patients.
Trisomy 18 (Edwards Syndrome)
Virtually all of the 1 in 6000 live-born infants with trisomy 18, or Edwards syndrome, have congenital heart defect (CHD), mainly ventricular septal defect and polyvalvular disease lesions.18 Valve malformations are characterized by redundant or thick myxomatous leaflets, long chordae tendineae, and hypoplastic papillary muscles.18 50% of these infants die in the first week of life, and only 5% to 10% survive beyond 1 year of age, albeit with significant developmental delay (see Chapter 174). CHD does not seem to affect survival in this population.19 Comparative fluorescence in situ hybridization (FISH) analysis of individuals with partial duplications of the long arm of chromosome 18 (< 5% of all cases) has shown that no single trisomy 18 critical region exists; rather, both proximal and distal regions of 18q appear to be required for full expression of the Edwards syndrome phenotype.20
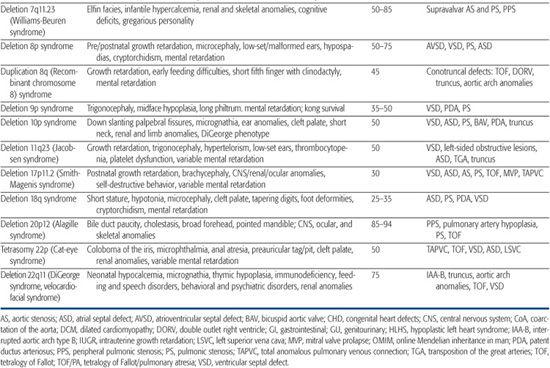
Table 481-1. Chromosomal Disorders Associated with Congenital Heart Defects (CHD)
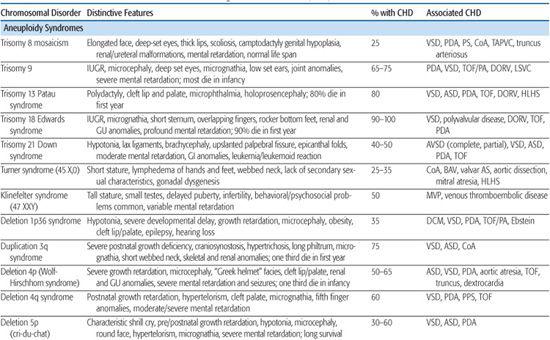
Trisomy 13 (Patau Syndrome)
Trisomy 13 is associated with a high incidence of congenital heart defect (CHD; 80%), infant mortality (> 90%), and severe developmental delay among those surviving infancy (see Chapter 174). Incidence is 1/10,000 to 1/15,000 live births. The most commonly associated cardiac lesions are atrial septal defect, ventricular septal defect, and patent ductus arteriosus.21,22 Valvular disease similar to that seen in trisomy 18 is also reported.22
Turner Syndrome
Turner syndrome (TS), or monosomy X, has a prevalence of 1/2500 to 1/5000 live births (see Chapter 174). It is associated with congenital heart defect (CHD) in at least 25% to 30% of patients. Left-sided obstructive lesions predominate, with bicuspid aortic valve (BAV) seen in up to 30% of subjects.23 Coarctation of the aorta is present in 10% to 20% of TS patients, and a more recent National Institutes of Health (NIH) study using magnetic resonance angiography revealed elongation of the transverse aortic arch, or pseudocoarctation, in 50% of TS patients.23 Other lesions such as partial anomalous pulmonary venous connection, atrial septal defect, ventricular septal defect, and persistent left superior vena cava are seen in 1% to 13% of patients.23 Individuals with TS are at increased risk of aortic root dilation, dissection, and sudden death,26 and although this occurs more typically in older adolescents and adults, aortic rupture has been reported in patients as young as 4 years.27 Whether this is solely attributable to the increased prevalence of hypertension, BAV, and aortic coarctation in this population, or rather indicates a diffuse vasculopathy in TS patients, is presently unclear.28
Other lesions such as partial anomalous pulmonary venous connection, atrial septal defect, ventricular septal defect, and persistent left superior vena cava are seen in 1% to 13% of patients.23 Individuals with TS are at increased risk of aortic root dilation, dissection, and sudden death,26 and although this occurs more typically in older adolescents and adults, aortic rupture has been reported in patients as young as 4 years.27 Whether this is solely attributable to the increased prevalence of hypertension, BAV, and aortic coarctation in this population, or rather indicates a diffuse vasculopathy in TS patients, is presently unclear.28
Current guidelines for CHD screening of patients diagnosed with TS include referral to a pediatric cardiologist for evaluation and echocardiographic imaging, followed by cardiac MRI at an age when the study may be performed without sedation (or earlier, if further imaging of the aortic valve or aorta is indicated on clinical grounds).29 In addition to screening for structural heart defects, ongoing blood pressure monitoring for hypertension is required, as well as ECG monitoring given an increased incidence of conduction and repolarization abnormalities, including prolongation of the QTc interval.30
The pathogenesis of CHD in monosomy X is unknown. No CHD critical region has emerged from phenotype-karyotype analyses. The association of CHD with fetal lymphedema32-34 has led to speculation that abnormal lymphatic and cardiovascular development are linked in this disorder. Interestingly, the gene for vascular endothelial growth factor (VEGF)-D, one of a family of growth factors involved in lymphatic and vascular development and permeability, maps to Xp22.1.
 DELETION/DUPLICATION SYNDROMES
DELETION/DUPLICATION SYNDROMES
Deletion/duplication syndromes usually result in multiple congenital anomalies; however, the spectrum of defects can vary significantly between patients, as can the exact size of the deleted or duplicated region. Two of the most common deletion syndromes associated with congenital heart defect (CHD) are discussed next, and a more complete list can be found in Table 481-1.
22q11 Deletion Syndrome
22q11 deletion syndrome is the most common chromosomal microdeletion syndrome with an incidence of 1 in 6000 live births. It encompasses a number of clinical entities previously known as DiGeorge syndrome, velocardiofacial syndrome, and cleft palate, absent thymus, congenital heart disease syndrome (CATCH-22), all of which share a ∼3-Mb deletion of 22q11.2 and variable phenotypic features.35-37 Seventy-five to 80% of 22q11 deletion syndrome patients have congenital heart defect (CHD),38,39 most commonly, tetralogy of Fallot, type B interrupted aortic arch, truncus arteriosus, perimembranous ventricular septal defect, and aortic arch anomalies (right aortic arch, high cervical arch, abnormal branching pattern).40-42 Conversely, the 22q11 deletion syndrome is a frequent finding among patients with certain types of CHD, especially type B interrupted arch (50%) and truncus arteriosus (35%). Studies have shown that, for certain defects, 22q11 deletion patients have higher operative mortality than nondeleted patients with the same lesion.43,44 Accordingly, the American Heart Association45 and the American Academy of Pediatrics (AAP)46 have more recently advocated fluorescence in situ hybridization (FISH) testing for the 22q11 deletion in all infants with the following cardiac lesions: type B interrupted arch,47,48 ventricular septal defect with aortic arch anomaly,49 truncus arteriosus,41,47,50-52 isolated aortic arch anomaly,53 and tetralogy of Fallot,49,54,55 especially when associated with either absent pulmonary valve, aortic arch or pulmonary artery anomalies, or aortopulmonary collaterals.41,47-55 In addition, testing is recommended for infants with any CHD and an additional feature of the 22q11 deletion syndrome, as well as any previously untested child/adolescent with the previously listed CHD and an additional feature of the syndrome.
Approximately 90% of patients with the DiGeorge syndrome phenotype will test positive by FISH for the microdeletion,56 thus indicating that other genetic (chromosome 10p deletion)57 and environmental causes (maternal diabetes mellitus)58 exist for this syndrome. Of FISH-positive individuals, 6% to 28% inherit the 22q11 deletion in an autosomal-dominant fashion,59 often from a parent not previously been diagnosed as having the 22q11 deletion syndrome. Subsequent analysis often reveals subtle syndromic features in the parent bearing the deletion,59 emphasizing that the 22q11 deletion phenotype can vary widely, even within families. For that reason, routine testing of parents of 22q11 deletion syndrome patients is recommended in order to provide crucial genetic counseling information to the family.
In the past decade, significant progress has been made toward understanding how haploinsufficiency for the ∼30 genes in the deleted region of 22q11 results in the cardiovascular and other phenotypic manifestations of the 22q11 deletion syndrome. A variety of mouse studies  suggest that Tbx1 is a major genetic determinant of the 22q11 deletion syndrome; however, there are likely other genes both within the deleted region and elsewhere whose alterations contribute to the observed variability in clinical presentation. Indeed, more recent studies have implicated vascular endothelial growth factor (VEGF) in such a modulatory role.64
suggest that Tbx1 is a major genetic determinant of the 22q11 deletion syndrome; however, there are likely other genes both within the deleted region and elsewhere whose alterations contribute to the observed variability in clinical presentation. Indeed, more recent studies have implicated vascular endothelial growth factor (VEGF) in such a modulatory role.64
7q11 Deletion Syndrome (Williams-Beuren Syndrome)
Williams-Beuren syndrome is an autosomal-dominant contiguous gene deletion syndrome. Incidence is approximately 1/20,000 live births. Cardiac defects affect 55% to 80% of Williams syndrome patients and most commonly include supravalvar aortic stenosis, supravalvar pulmonic stenosis, and peripheral pulmonic stenosis.65,66 In general, pulmonary vessel stenosis improves over time, whereas aortic vessel stenosis tends to progress.65,67 Histologically, the narrowed vessels are characterized by medial disorganization and intimal hypertrophy.68 This arteriopathy can affect other vessels, most notably the renal arteries, where it can lead to hypertension, or the coronary arteries, where it has been implicated in rare sudden death.69 Other lesions such as valvar aortic stenosis, aortic hypoplasia, and coarctation have also been reported.
A submicroscopic deletion of chromosome 7q11.23 is detectable by fluorescence in situ hybridization (FISH) in approximately 90% of patients with the clinical diagnosis of Williams syndrome, and the deletion usually arises de novo.70,71 The deleted region encompasses 25 to 30 genes, including the STX1A,72 elastin (ELN),71-73 and LIMK174 genes. The observed clinical phenotype depends on which genes are deleted. Heterozygous loss of the elastin gene is believed to cause the elastic arteriopathy that characterizes the cardiovascular manifestations of Williams syndrome71 because deletions solely within the elastin gene cause isolated supravalvar aortic and/or pulmonic stenosis.75 Deletions of other genes within the region have been linked to the craniofacial76 and neurocognitive77 defects. Given the clinical variability of the syndrome and the potential benefit of early diagnosis and treatment of associated problems such as hypercalcemia, hypertension, and learning disabilities, FISH testing for the 7q11.23 deletion should be considered for patients with supravalvar aortic or pulmonic stenosis, or with peripheral pulmonic stenosis that persists beyond infancy.
 CONGENITAL HEART DEFECT ASSOCIATED WITH SINGLE GENE DISORDERS
CONGENITAL HEART DEFECT ASSOCIATED WITH SINGLE GENE DISORDERS
For information on congenital heart defect (CHD) associated with single gene disorders, see Table 481-2.
Noonan Syndrome
Noonan syndrome is an autosomal-dominant disorder characterized by dysmorphic features (hypertelorism, ptosis, epicanthal folds, low-set posteriorly rotated ears), short stature, and congenital heart defect (CHD). It occurs with an estimated frequency of 1/1000 to 1/2500 live births, making it the most common nonchromosomal syndrome associated with CHD. CHD are present in 80% to 90% of Noonan syndrome patients, most often valvar pulmonic stenosis and hypertrophic cardiomyopathy.78,79
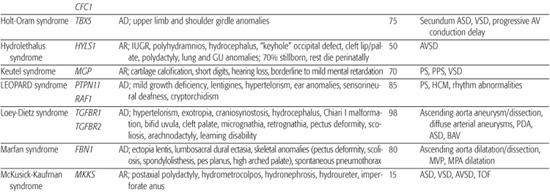
Table 481-2. Single Gene Disorders Associated with Syndromic Congenital Heart Defects (CHD)
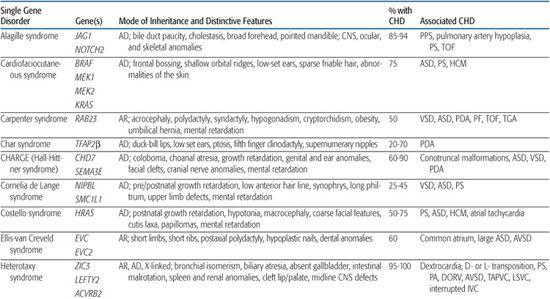

After Noonan syndrome was mapped to chromosome 12q24, a candidate gene approach led to the discovery that missense mutations in the PTPN11 gene cause the syndrome.80PTPN11 encodes the protein tyrosine phosphatase SHP-2, which functions in the RAS-mitogen activated protein kinase (MAPK) pathway and is implicated in semilunar valvulogenesis.81PTPN11 mutations account for roughly half of clinically diagnosed Noonan syndrome, indicating that Noonan syndrome is genetically heterogeneous. More recent studies have revealed mutations in KRAS,82SOS,81,83 or RAF,84,85 genes that encode other proteins in the RAS-MAPK pathway, in a significant percentage of Noonan syndrome patients that lack PTPN11 mutations. Noonan syndrome patients harboring PTPN11 or SOS mutations are much more likely to have pulmonic stenosis (70%) than hypertrophic cardiomyopathy (< 10%).81,86 Conversely, 80% to 95% of individuals with mutations in the CR2 domain of RAF exhibit hypertrophic cardiomyopathy, compared to an 18% incidence among Noonan syndrome patients in general.84,85 All disease-causing mutations in Noonan syndrome are believed to exhibit gain-of-function behavior, indicating that the pathobiologic basis for the developmental anomalies seen in Noonan syndrome are caused by dysregulation of the RAS-MAPK pathway.87
Mutations in PTPN11, KRAS, RAF, and other genes in the RAS-MAPK pathway have more recently been shown to cause a variety of disorders that overlap phenotypically with Noonan syndrome. 
Clinical testing for most, if not all, of the mutated RAS-MAPK genes is now available. However, the genetic heterogeneity of most of the Noonanlike disorders limits the usefulness of these tests because failure to detect a mutation cannot exclude the diagnosis. These syndromes are typically caused by sporadic mutations, although familial inheritance is reported for Noonan syndrome.
Alagille Syndrome
Alagille syndrome is an autosomal-dominant disorder consisting of bile duct paucity, cholestasis, typical facies, and cardiovascular, skeletal, and ocular anomalies (see Chapter 421). The incidence is estimated at 1/70,000 live births, and approximately half result from de novo mutations. Congenital heart defect (CHD) is present in more than 90% of Alagille patients, most commonly, right-sided lesions such as peripheral pulmonary stenosis or hypoplasia, pulmonary valve stenosis, and tetralogy of Fallot.100 Left-sided lesions and septal defects are less common.100 Noncardiac vascular anomalies such as aneurysms of the internal carotid, basilar, or middle cerebral artery; renal artery stenosis; and moyamoya disease have been reported as a cause of mortality in individuals with Alagille syndrome.101
More than 90% of individuals with Alagille syndrome harbor a mutation in the JAG1 gene102-104 located on chromosome 20p12. JAG1 encodes a cell surface protein that acts as a ligand in the Notch signaling pathway. Most JAG1 mutations are believed to result in haploinsufficiency, and no clear genotype-phenotype correlations have emerged with respect to type or severity of CHD.100 Mutations in the NOTCH2 gene, which encodes a Notch receptor believed likely to interact with Jagged1106 have been found in less than 1% of patients with Alagille syndrome.107 Hence, the Notch signaling pathway, which is involved in cell fate determination and is required for normal embryonic development in the mouse,108 appears to play a central role in the pathogenesis of Alagille syndrome. However, the wide variability in clinical presentation of the syndrome, even among family members harboring the same mutation, suggests that significant genetic and/or environmental modifiers have yet to be uncovered.
Patients suspected to have Alagille syndrome should be considered for JAG1 mutational analysis, followed by cytogenetic testing if no intragenic JAG1 mutation is detected. Testing should be considered for patients with a strong family history of right-sided lesions who do not exhibit extracardiac manifestations of the syndrome.109,110 Identification of JAG1 mutations in patients with suspected Alagille syndrome should prompt testing of appropriate family members.
Holt-Oram Syndrome
Holt-Oram syndrome is a highly penetrant but variably expressive autosomal-dominant disorder characterized by upper limb deformities often accompanied by congenital heart defect (CHD).111 The incidence is approximately 1/100,000 live births, and the vast majority are believed to arise from new mutations.112 CHD is present in 75%, typically secundum atrial septal defect, muscular ventricular septal defect, or progressive atrioventricular conduction delay, singly or in combination.113,114
Genetic linkage analysis of affected families led to the discovery that Holt-Oram syndrome is caused by mutations in the T-box transcription factor gene TBX5 on chromosome 12q24.1.116,117 When strict diagnostic criteria are used, approximately 75% of individuals with Holt-Oram syndrome will have TBX5 coding region changes,118 mostly nonsense or frameshift mutations predicted to result in haploinsufficiency. The remaining one fourth are believed to have noncoding region changes or deletions rather than mutations at other loci. Specific missense mutations in TBX5 that result in Holt-Oram syndrome have been shown to abrogate its binding to the transcription factor GATA4, and mutations in GATA4 that disrupt interaction with TBX5 also result in septal defects in humans.119 The molecular mechanism underlying the conduction defects in Holt-Oram syndrome is unknown. 
Ellis-van Creveld Syndrome
Ellis-van Creveld syndrome is a relatively rare autosomal-recessive skeletal dysplasia with congenital heart defect (CHD) in ∼60% of patients. Large atrial septal defect, common atrium, and atrioventricular septal defect are the most commonly reported lesions.121
Linkage analysis and positional cloning studies identified mutations in two unrelated genes on chromosome 4p16 as causes of Ellis-van Creveld syndrome.122-124 It has been estimated that mutations in the novel genes EVC and EVC2 account for two thirds of Ellis-van Creveld patients, suggesting that there may be further genetic heterogeneity.125 Mutations in either gene can give rise to the full spectrum of clinical phenotypes, and most mutations are believed to result in loss of function. Although the functions of the proteins encoded by EVC and EVC2 are unknown, both are expressed in the developing heart and skeleton. 
CHARGE Syndrome (Hall-Hittner Syndrome)
CHARGE syndrome (coloboma, heart anomaly, atresia choanal, retardation of mental and somatic development, genital and ear anomalies) was first described as a nonrandom association of congenital anomalies by Hall127 and Hittner128 et al. Over time, other abnormalities such as cranial nerve anomalies (weak chewing/sucking, facial weakness, sensorineural hearing loss, and swallowing problems)129 and facial clefts have been recognized as major features of CHARGE syndrome, and diagnostic criteria have been revised and updated accordingly.130,131 Incidence is estimated at 1/10,000 to 1/15,000 live births, with most patients representing new mutations. Coronary heart defect (CHD) remains a minor diagnostic sign and is observed in 60% to 90% of patients. Conotruncal malformations are most commonly seen, followed by septal defects, patent ductus arteriosus, aortic arch anomalies, and left heart lesions.132,133
Identification of a microdeletion on chromosome 8q12 by array comparative genomic hybridization in a patient with CHARGE syndrome led to the discovery that mutations in the chromodomain helicase DNA-binding gene (CHD7) are responsible for approximately two thirds of clinically diagnosed patients with the syndrome.133-136 Most mutations are believed to result in loss of expression, or haploinsufficiency. Mutational analysis has confirmed rare instances of autosomal-dominant inheritance. No strong genotype-phenotype correlations have emerged; indeed, there are reports of differences in clinical presentation between sib pairs with identical mutations.135 The CHD family of genes is involved in chromatin remodeling and regulation of gene expression during development. CHD7 is expressed highly in embryonic neural crest, consistent with the spectrum of defects in neural crest–derived tissues seen in CHARGE syndrome patients.
Char Syndrome
Char syndrome is an autosomal-dominant disorder characterized by facial dysmorphism (duck-bill lips, ptosis, and low-set ears), hand anomalies, and patent ductus arteriosus (PDA).138
Heterotaxy Syndromes
Heterotaxy (from the Greek “heteros” meaning “other” and “taxy” meaning “order” or “arrangement”) is a malformation complex characterized by loss of normal orientation of thoracoabdominal organs with respect to the left-right body axis. “Situs solitus” refers to the usual arrangement of the heart and abdominal organs, whereas “situs inversus” implies the complete reversal, or mirror image. Any other arrangement is considered “situs ambiguous” or heterotaxy. The incidence of congenital heart defect (CHD) in situs inversus is 3% to 9%,141 whereas virtually all patients with heterotaxy have complex CHD, as well as abnormalities of other organs (Table 481-2). 
Heterotaxy is a clinically and genetically heterogeneous disorder with an incidence of 1/10,000 live births.144 Most examples are sporadic, but familial recurrence is well documented and has led to the mapping and identification of a number of loci involved in left-right asymmetry. Analysis of kindreds with an X-linked form of heterotaxy led to the discovery that mutations in the putative zinc-finger transcription factor ZIC3 can cause both familial and sporadic examples of the syndrome.145 Within these kindreds, phenotypes ranged from multiple organ system involvement to isolated CHD,146 suggesting that a wide spectrum of disease expression exists. Using a candidate approach that involves searching for alterations in genes implicated in left-right axis determination in model organisms, a small number of heterotaxy patients harboring mutations in CFC1,147LEFTY A,148 and ACVR2B149 have been identified. Whether these mutations account for a significant portion of heterotaxy syndrome awaits further study. 
Marfan Syndrome
Marfan syndrome is an autosomal-dominant disorder of connective tissue caused by mutations in the fibrillin-1 gene FBN1.150 It is characterized by skeletal, ocular, and cardiovascular manifestations that exhibit complete penetrance but variable expression (see Chapter 179).
Table 481-3. Single Gene Disorders Associated with Isolated Congenital Heart Defects
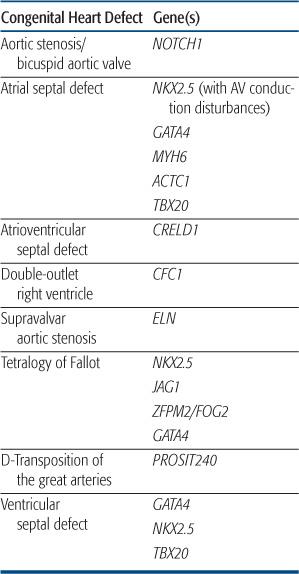
 CONGENTIAL HEART DEFECT ASSOCIATED WITH NONSYNDROMIC SINGLE GENE DISORDERS
CONGENTIAL HEART DEFECT ASSOCIATED WITH NONSYNDROMIC SINGLE GENE DISORDERS
Congenital heart defect (CHD) most often occurs as an isolated defect rather than as part of a syndrome. Although the etiology of most nonsyndromic CHD is likely multifactorial, more recent studies have shown that mutations in single genes can result in specific forms of CHD in a small proportion of patients. However, the same mutation can result in different forms of CHD even within the same kindred, reinforcing the idea that additional genetic and/or environmental factors contribute to expression of the CHD phenotype. Table 481-3 contains a list of single gene disorders and the CHD with which they have been associated. 
NKX2.5
The first discovery of single gene mutations resulting in nonsyndromic cardiac defects came from analysis of several families with autosomal-dominant congenital heart defect (CHD) consisting most commonly of atrial septal defect and atrioventricular conduction delay.158 There was progressive conduction disease in some individuals, and a strong history of sudden death in these families. After linkage studies mapped the disease locus in these kindred to chromosome 5q35, mutations were found in the candidate gene NKX2.5, a homeobox transcription factor known to be involved in cardiac development from animal studies.158 These mutations were found in all affected individuals, were absent in unaffected family members and other control samples, and were shown to alter the DNA-binding and/or transcriptional activation function(s) of the protein.159 Subsequent studies extended the association between NKX2.5 mutations, CHD, and atrioventricular conduction disorders,160 and animal models have confirmed a role for NKX2.5 in maintenance of the atrioventricular conduction system.161 Alterations in the NKX2.5 coding region have also been found in a small proportion of patients with sporadic atrial septal defect (1%–4% of patients),162 as well as other lesions, including ventricular septal defect, tetralogy of Fallot, and hypoplastic left heart syndrome.163,164 However, the functional significance of some of these mutations awaits further study. Testing for NKX2.5 mutations is available clinically and may be useful in familial atrial septal defect to predict those at risk for progressive atrioventricular conduction disorders.
GATA4
GATA4 is transcription factor that partners with NKX2.5 and has been shown in animal models to regulate expression of genes critical to myocardial differentiation and function. Mutations in GATA4 were reported in two families with congenital heart defect (CHD) consisting of atrial septal defect in all, with some individuals having a second defect (ventricular septal defect, atrioventricular septal defect, pulmonic stenosis).119 There were no associated conduction disorders.  Subsequent studies have shown GATA4 sequence alterations in a small percentage of sporadic instances of atrial septal defect, as well as tetralogy of Fallot and atrioventricular septal defect.165,166
Subsequent studies have shown GATA4 sequence alterations in a small percentage of sporadic instances of atrial septal defect, as well as tetralogy of Fallot and atrioventricular septal defect.165,166
NOTCH1
Analysis of several families with autosomal-dominant aortic valve disease led to the discovery of mutations in the signaling molecule NOTCH1 in individuals with bicuspid aortic valve and aortic stenosis.167 Affected family members, including several with trileaflet aortic valves, developed severe valve calcification. Animal models confirmed that NOTCH1 is highly expressed in the developing aortic valve, and may play a role in regulating calcium deposition.167
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


