GENETIC COUNSELING
Key Points
• All pregnant women should receive a basic genetic risk evaluation and assessment as part of prenatal care.
• All pregnant women should be offered serum analyte screening for fetal aneuploidies, neural tube defects (NTD), and von Willebrand Disease (VWD). This may be combined with ultrasound evaluation of fetal nuchal translucency and nasal bone if available. Cell-free DNA testing for fetal aneuploidy should be offered in specific circumstances of increased genetic risk.
• Invasive testing for fetal aneuploidies and chromosomal deletion syndromes by amniocentesis, chorionic villus sampling (CVS), or cord blood sampling should be offered to women who have a risk that meets or exceeds specific criteria.
• Follow-up counseling and care should be available for any woman with abnormal karyotype results or evidence of fetal anomalies that are significant or incompatible with life.
• Women with inherited genetic abnormalities or diseases should be offered fetal testing if it is possible and should receive appropriate medical management and prenatal care.
Background
Our understanding of the scientific basis of genetics has advanced with increasing rapidity over the past century and a half, since Gregor Mendel’s groundbreaking studies of the pea plant. With the definition of the human genome, genetics has assumed a key position in clinical medicine. Many of our future diagnostic and therapeutic options will be based on or derived from the principles of genetics, making it important for clinicians to have a thorough understanding of the science (1).
Before the explosion of information in molecular genetics, the terminology of clinical genetics was based in the concepts of the gene as the basic, smallest, unit of inheritance. An understanding of the definitions cogent to these clinical terms remains important in communications regarding clinical genetics; they should be clearly understood. Some of the terms that should be familiar are genetics, heredity, counseling, phenotype, genotype, gene, allele, locus, trait, autosomal, sex-linked, heterozygote, homozygote, pedigree, crossing-over, and syndrome (2).
MENDELIAN INHERITANCE
Background
Single-gene inheritance follows the mendelian principles of unitary inheritance, allelic segregation, and independent assortment. It is elucidated by following parents and their progeny and observing distinct patterns emerge: A specific locus is passed, intact, to the offspring. The gene is passed by each of the parents. Therefore, there are two copies. When the appropriate number of copies is present in an individual, an abnormal trait may be identified. This is the basic concept; however, there are variations on this theme (3).
Evaluation
Genetics
• Autosomal dominant: These genetic traits are inherited in the presence of only one dominant gene.
• More than half of the approximately 5000 mendelian conditions behave in an autosomal dominant fashion.
• The specific phenotype appears in every generation, and every affected individual has an affected parent (with exceptions discussed later in this chapter).
• A child of an affected parent has a 50% chance of inheriting the abnormal gene, and, conversely, the phenotypically normal family member does not transmit the phenotype to his or her offspring.
• Men and women are equally likely to pass the phenotype to their children of either sex.
• Apparent exceptions to these rules are either the result of new mutations in the germ cells (sperm and ova) or situations in which, despite the presence of the abnormal gene, the disease is not expressed in the individual.
• Two copies of abnormal dominant genes (homozygosity) are generally lethal.
• Common examples of autosomal dominant disorders are achondroplasia, Huntington disease, Marfan syndrome, and neurofibromatosis (4).
• Autosomal recessive: These genetic traits require the presence of two paired genes for expression of the abnormality. These traits are generally biochemical abnormalities.
• An autosomal recessive disease is not seen more than once in a pedigree, outside of the siblings of an affected member of the family; the risk of occurrence of the disease for each sibling of the affected individual is 25%.
• An unaffected sibling of an affected individual has a two-thirds chance of being a carrier (heterozygote).
• When the gene is rare, the parents of an individual with such an autosomal recessive condition will be close relatives of each other.
• Male and female offspring are equally likely to be affected.
• Autosomal recessive disorders (and some sex-linked recessive disorders) involving single biochemical changes, used to be referred to as “inborn errors of metabolism.” Examples are Tay-Sachs disease, phenylketonuria (PKU), and maple syrup urine disease.
• Sex-linked dominant and recessive
• The dominant forms require only a single gene on the X chromosome to express the abnormality.
• The recessive forms require only one gene on the X chromosome in males but one gene on each of two X chromosomes in females.
 The incidence of X-linked recessive conditions is much more common in male than in female offspring.
The incidence of X-linked recessive conditions is much more common in male than in female offspring.
 Phenotypically normal daughters of affected men (the recessive form) have a 50% chance of passing the abnormal gene to their sons, who will demonstrate the trait. An affected man, however, never has an affected son.
Phenotypically normal daughters of affected men (the recessive form) have a 50% chance of passing the abnormal gene to their sons, who will demonstrate the trait. An affected man, however, never has an affected son.
 Heterozygous women (carriers of the recessive form) are generally unaffected.
Heterozygous women (carriers of the recessive form) are generally unaffected.
 In the more rare X-linked dominant conditions, affected men have no affected sons and no normal daughters. Both male and female offspring of female carriers have a 50% chance of inheriting the phenotype.
In the more rare X-linked dominant conditions, affected men have no affected sons and no normal daughters. Both male and female offspring of female carriers have a 50% chance of inheriting the phenotype.
• Not all women who inherit either a single dominant gene or a pair of recessive genes will phenotypically express their genotype, as explained by the Lyon hypothesis (see discussion below) (5).
• Factors influencing the phenotype in mendelian inheritance–based genotypes:
• Penetrance: Occasionally, an individual with a specific inherited trait or disease (especially autosomal dominant) may not demonstrate the typical phenotype. This may be secondary to the age of the individual (dies before manifestation of the phenotype) or the influence of another gene or the environment. This is referred to as diminished penetrance, where there is a lower-than-expected incidence of the phenotype in the genotypic population.
• Expressivity: When the phenotype varies in the same genotype (not so much in the same family, but in other families), the condition is considered to have greater variability of expression. This can vary from minor to severe expressions and may be due to sex-influenced or limited expression. In some autosomal dominant disorders, the phenotypic manifestation differs between males and females.
• Anticipation: The term for the situation when a dominant condition appears to present at a younger age in each subsequent generation or when the severity of the disease or trait is increased with each subsequent generation. This may be related to conditions where there is additional expansion of triplets in the genetic code, which is discussed later in this chapter.
• The Lyon hypothesis: This hypothesis holds that whenever there is more than one X chromosome in the genome, one may be randomly inactivated, existing as inactive chromatin (the Barr body). Since the process is random, the percentage of cells with one or the other active X chromosome will vary within specific organ systems and within the individual as a whole. If an abnormal allele is on one of the X chromosomes, its affect may either be unopposed or absent in a percentage of cells, creating a situation in which a heterozygote may express the abnormal allele to varying degrees. It is now believed that this effect is caused by an X-inactivation–specific transcript gene on the long arm of the X chromosome (5,6).
• Pleiotropy: Although each abnormal gene has one direct effect for which it is identified, there may be secondary or tertiary effects from that same gene on other aspects of an individual’s physiology. This has to do with different characteristics of use of the RNA product and subsequent protein product of the gene. Each of these phenotypic effects is believed to be the pleiotropic result of an abnormal protein as transcribed and translated from the specific gene. Pleiotropy may be seen in Marfan syndrome and in the abnormalities of the fibroblast growth factor gene.
• Codominance: Certain phenotypic traits demonstrate codominance, in which both traits are expressed, either from alleles in the same locus or from alleles at separate loci dictating a phenotypic appearance. Examples are hemoglobinopathies and blood types.
• A compilation of information on mendelian inherited disorders may be found updated with references online: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db = OMIM (7).
Diagnosis
Clinical Manifestations
• Chromosomal disorders
• General rules
 Chromosomes can be seen only during the stage of the cell cycle when the genetic material within the nucleus condenses to become microscopically (cytogenetically) visible.
Chromosomes can be seen only during the stage of the cell cycle when the genetic material within the nucleus condenses to become microscopically (cytogenetically) visible.
 New staining techniques create bands on chromosomes. Each technique develops a distinctive banding pattern so that minute areas of the chromosome may be visibly distinguished and compared. With high-resolution karyotyping, chromosomes can be subdivided into hundreds of subbands, with an average length of about 3.5 million base pairs (8).
New staining techniques create bands on chromosomes. Each technique develops a distinctive banding pattern so that minute areas of the chromosome may be visibly distinguished and compared. With high-resolution karyotyping, chromosomes can be subdivided into hundreds of subbands, with an average length of about 3.5 million base pairs (8).
 One set of 23 chromosomes is inherited from each parent. A pair of chromosomes is called a homolog (e.g., the two number 1 chromosomes). They have a similar cytogenetic appearance and carry “matching” genetic material down to the locus of each gene and the DNA code itself.
One set of 23 chromosomes is inherited from each parent. A pair of chromosomes is called a homolog (e.g., the two number 1 chromosomes). They have a similar cytogenetic appearance and carry “matching” genetic material down to the locus of each gene and the DNA code itself.
 The two types of cell division, mitosis and meiosis, result in the daughter cells with the appropriate number of chromosomes and the correct amount of genetic material. Mitosis results in two cells with equal genetic material. Meiosis results in four gametes in men and one in women, each with a haploid complement of chromosomes (the other three gametes in women are usually lost).
The two types of cell division, mitosis and meiosis, result in the daughter cells with the appropriate number of chromosomes and the correct amount of genetic material. Mitosis results in two cells with equal genetic material. Meiosis results in four gametes in men and one in women, each with a haploid complement of chromosomes (the other three gametes in women are usually lost).
 Chromosomal disorders are primarily the result of a deficiency or excess of a significant amount of genetic material.
Chromosomal disorders are primarily the result of a deficiency or excess of a significant amount of genetic material.
 The presence of an extra chromosome or chromosome segment can result in a distinctive genetic condition or a peculiar phenotype.
The presence of an extra chromosome or chromosome segment can result in a distinctive genetic condition or a peculiar phenotype.
 Collectively, chromosome disorders are more common than are single-gene disorders and affect 6 to 10 out of 1000 liveborn infants.
Collectively, chromosome disorders are more common than are single-gene disorders and affect 6 to 10 out of 1000 liveborn infants.
 Aneuploidies are chromosomal disorders relating to abnormal numbers of chromosomes. One extra chromosome is called a trisomy (three of one chromosome), and one less chromosome is called a monosomy (one of the chromosome instead of two). An additional set of 23 chromosomes is referred to with the suffix of “ploidy” (69 chromosomes, triploidy; 92 chromosomes, tetraploidy).
Aneuploidies are chromosomal disorders relating to abnormal numbers of chromosomes. One extra chromosome is called a trisomy (three of one chromosome), and one less chromosome is called a monosomy (one of the chromosome instead of two). An additional set of 23 chromosomes is referred to with the suffix of “ploidy” (69 chromosomes, triploidy; 92 chromosomes, tetraploidy).
 Rearrangements (recombination of the genetic material) occur both in somatic mitosis and gametic meiosis. Cell division errors can and do occur. From a cytogenetic standpoint, the most error-prone step in meiosis is anaphase I of the first division of meiosis. When the chromosomes do not segregate to the opposite poles of the dividing cell, the resulting gametes can have an extra chromosome or be deficient for the nonsegregating chromosome. This is called nondisjunction. Fertilization of these gametes produces an individual with an abnormal number of chromosomes (Fig. 29-1).
Rearrangements (recombination of the genetic material) occur both in somatic mitosis and gametic meiosis. Cell division errors can and do occur. From a cytogenetic standpoint, the most error-prone step in meiosis is anaphase I of the first division of meiosis. When the chromosomes do not segregate to the opposite poles of the dividing cell, the resulting gametes can have an extra chromosome or be deficient for the nonsegregating chromosome. This is called nondisjunction. Fertilization of these gametes produces an individual with an abnormal number of chromosomes (Fig. 29-1).
 Abnormalities in the arms of the chromosomes (i.e., deletions, additions, inversions) are referred to by the letters p for the short arm and q for the long arm of the chromosome. Such abnormalities and crossing-over of genetic materials generally occur during meiosis in the germ cells. This is demonstrated at http://www.accessexcellence.org/RC/VL/GG/comeiosis.html (Accessed March 29, 2013).
Abnormalities in the arms of the chromosomes (i.e., deletions, additions, inversions) are referred to by the letters p for the short arm and q for the long arm of the chromosome. Such abnormalities and crossing-over of genetic materials generally occur during meiosis in the germ cells. This is demonstrated at http://www.accessexcellence.org/RC/VL/GG/comeiosis.html (Accessed March 29, 2013).
 Chromosomal terminology is established by international conferences and published on a regular basis. Some basic terminology is needed to interpret laboratory reports (9).
Chromosomal terminology is established by international conferences and published on a regular basis. Some basic terminology is needed to interpret laboratory reports (9).
• Chromosomal syndromes
 Aneuploidy: abnormal numbers of chromosomes
Aneuploidy: abnormal numbers of chromosomes
– Monosomy: 45 chromosomes
∗ Autosomal monosomy is generally lethal.
∗ X chromosome monosomy is referred to as Turner syndrome.
– Trisomy: 47 chromosomes
∗ Trisomy 21 is Down syndrome. It has an incidence of 1 in 800 to 1000 newborns. Ninety-five percent of cases of Down syndrome result from maternal nondisjunction, and 5% result from translocations and mosaicisms.
∗ Trisomy 18 is Edwards syndrome. It has an incidence of 1 in 8000 newborns. Eighty-five percent of fetuses with this syndrome die between 10 and 40 weeks of gestation.
∗ Trisomy 13 is Patau syndrome. This syndrome affects 1 in 20,000 newborns. Median survival is 3 days, and 90% die by 1 month of age.
∗ XXY is Klinefelter syndrome. This genetic disorder gives rise to infertile, tall males.
∗ XXX syndrome: Females with variable intelligence; some develop mental problems.
∗ XYY syndrome: Tall phenotypically normal males, with learning disabilities and aggressiveness.
∗ Other trisomies are rare except as mosaicisms or partial trisomies secondary to additions.
 Polyploidy
Polyploidy
– Partial hydatidiform mole (69 chromosomes) occurs when the extra set of chromosomes is paternal.
– Intrauterine growth restriction of fetus and placenta (69 chromosomes) occurs when the extra set is of maternal origin.
– Higher number ploidies are generally lethal.
 Deletions and additions; copy number variations (CNV)
Deletions and additions; copy number variations (CNV)
– Deletion of 4p is Wolf-Hirschhorn syndrome.
– Deletion of 5p is cri-du-chat syndrome.
– Microdeletion of 22q11.2 is Shprintzen syndrome and DiGeorge syndrome, which are two extremes of the same deletions, or current methods are too inexact to further distinguish the deletion site. Note that Shprintzen syndrome is also known as velocardiofacial syndrome.
– Ring chromosomes are deletions at each end of the chromosome that “heal” with both ends stuck to each other.
– Deletions and additions are now generally detected in molecular genetics by microarray comparative genomic hybridization with fluorescence, probe amplification, or quantitative polymerase chain reaction (PCR) (10,11).
 Translocations
Translocations
– Reciprocal: Breaks at two points with transfer of material before healing.
– Balanced: Equal amounts of genetic materials are exchanged. One-quarter of gametes are normal, one-half are duplications and deletions, and one-quarter are translocation carriers.
– Robertsonian: Only occurs with acrocentric chromosomes 13, 14, 15, 21, and 23. Central fusion translocation is the most common form of translocation.
– Isochromosomes: Either two long arms (q) or two short arms (p) are fused into a new chromosome.
 Inversions
Inversions
– Breaks occur at two points, and the material inverts before healing.
– Paracentric: The breaks occur away from the centromeres and may be balanced or unbalanced.
– Pericentric: The breaks include the centromere.
 Mosaicisms
Mosaicisms
– There are two or more cell lines in the body made up of different chromosomal complements but from the same zygote (e.g., 46XX, 45X, the loss of an X chromosome during early mitotic divisions).
– The phenotype of the mosaic individual depends on the location and percentage of abnormal cells in the various organ systems.
– Mosaicism in the trophoblast and placenta is relatively common. – Gonadal mosaicism may explain spontaneous mutations occurring more frequently than expected in a sibship.
 Chimerism
Chimerism
– There are two cell lines in the body made up of different chromosomal complements from two different zygotes.
– Fraternal twins with exchange of hematopoietic stem cells may lead to an individual with two different DNAs on blood testing (11).
– Commonly seen in animals but exceedingly rare in humans, the fusion of two fertilized ova may cause an individual with two separate cell lines in some organ systems (12).
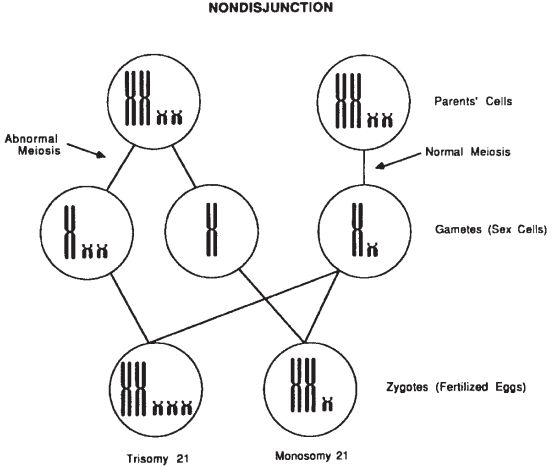
Figure 29-1. Nondisjunction resulting from an abnormal meiotic division yields an aneuploidy gamete and zygote (6).
NONMENDELIAN INHERITANCE
Background
A number of nonclassic patterns have been described that differ from the established patterns of inheritance. Developments in molecular genetics have allowed these unusual patterns of inheritance to be more clearly defined.
Evaluation
Genetics
• Triplet repeats in DNA: In this situation, a region of DNA exhibits repetitive repeats of a triplet nucleotide sequence (e.g., CGG at Xq27 in fragile X or Martin-Bell syndrome). During crossing-over in meiosis, sister chromatids may exchange some DNA, which may lengthen the section of the triplet nucleotide sequence. When this happens and the sequence attains a certain length, methylation may occur and damage or inactivate the DNA, causing the specific abnormal trait. Characteristic findings with this are as follows:
• Increasing severity of the abnormal trait with subsequent generations.
• Cytogenetic techniques are not preferable for detection.
• Restriction endonuclease digestion and Southern blot analysis for nucleotide triplets and DNA methylation are the preferred detection methods (13,14).
• This situation may explain some of the cases of penetrance and varied expressivity in phenotypes with the same genetic disorders.
• Examples of disorders associated with recurrent nucleotide sequences include fragile X syndrome, myotonic dystrophy, Huntington disease, Friedrich ataxia, X-linked spinal and bulbar muscular atrophy (Kennedy disease), spinocerebellar ataxia types 1 and 2, dentatorubral-pallidoluysian atrophy, and Machado-Joseph disease (2,16):
 Fragile X syndrome is transmitted as an X-linked disorder.
Fragile X syndrome is transmitted as an X-linked disorder.
– The number of CGG repeats varies in four classifications:
∗ Unaffected (less than 45 triplets)
∗ Intermediate (45 to 54 triplets)
∗ Premutation (55 to 200 triplets)
∗ Full mutation (greater than 200 triplets)
– The American College of Obstetricians and Gynecologists (ACOG) Committee on Genetics recommends that women with a family history of fragile X–related disorders, unexplained mental retardation or developmental delay, autism, or premature ovarian insufficiency are candidates for genetic counseling and fragile X premutation carrier screening (the FMR1 DNA mutation should also be performed after genetics counseling) (15).
• Mitochondrial inheritance: The mitochondrion, the site of oxidative phosphorylation within the somatic cell, carries its own genetic information.
• Mitochondrial DNA (mtDNA) exists as a circular chromosome within the mitochondrion.
• During cell division, the mitochondria are thought to be randomly distributed to the daughter cells.
• Variable expression is commonly seen in mitochondrial diseases because the phenotypic trait relates to the proportions of normal and abnormal mitochondria.
• Peculiar to mitochondrial inheritance is its maternal inheritance pattern. A mother transmits some of her mitochondria to her eggs and offspring. A father (through his sperm) transmits none. Recurrence risks can be substantial.
• Examples of mtDNA inheritance are myoclonic epilepsy with ragged red fibers, Leber hereditary optic neuropathy, Leigh syndrome, and pigmentary retinopathy (2,16).
• Imprinting: Some of the genetic code is inherited in a nonactive form from one parent and is only turned on or made active by a gene from the other parent. This appears to be sex specific in the offspring.
• The best studied demonstration of imprinting is in the Prader-Willi/Angelman syndrome complex. It is linked to a cytogenetically demonstrable or molecularly identified microdeletion in the long arm of chromosome 15. When inherited from the father, it is the Prader-Willi syndrome. Angelman syndrome has a completely different phenotype when inherited with the identical Prader-Willi microdeletion but from the mother (2,16).
• The parental origin of the genetic material appears to have a profound effect on clinical expression and phenotype (11).
• Imprinting exerts its effect on the phenotype essentially by controlling the dosage of gene effect, making a biallelic trait monoallelic in some situations. The reason for such a mechanism has not yet been elucidated.
• Uniparental disomy exists when both members of one pair of chromosomes are inherited from the same parent. This may occur from other mechanisms, but the most common mechanism may be “trisomic rescue.” Most often, there is no consequence of this phenomenon except for chromosomes 6, 7, 11, 14, and 15. Isodisomy is the term when both chromosomes in a pair are from the same single chromosome in a parental pair (3,13).
MULTIFACTORIAL AND POLYGENIC INHERITANCE
Background
Multifactorial disorders generally result from the complex interaction between genes and environment. The underlying cause may be compound, and the role of the environment may have a multifaceted involvement in the expression of the genetic material.
Evaluation
Genetics
• Multifactorial defects fail to show the classic mendelian inheritance patterns seen for single-gene defects; therefore, recurrence risks are primarily based on empirical data.
• Multifactorial defects are obviously familial, but there is no distinctive pattern of inheritance within a family (16).
• Risk to first-degree relatives is approximately the square root of the populational risk.
• The risk for second-degree relatives decreases sharply from that for first-degree relatives but declines less rapidly with more distant kinship.
• Recurrence risk is higher when more family members are affected.
• The more serious the defect, the higher the recurrence risk.
• Several loci are involved in creating the phenotypic trait.
• The loci act in concert in an additive fashion, each adding or subtracting a small amount from the phenotype.
• The environment interacts to varying degrees to produce the final phenotype (e.g., vitamin deficiency, excess hormone production).
• The frequency of similarly affected co-twins is higher among monozygotic than among dizygotic twins.
• A number of different conditions fall into the category of multifactorial inheritance, including cleft lip with or without cleft palate, isolated cleft palate, congenital dislocation of the hip, pyloric stenosis, isolated NTD (including anencephaly and spina bifida), and isolated congenital heart defects (recurrence risks of 2% to 4%) (Table 29-1).
• For these conditions, empirical data suggest that the recurrence risk is significantly higher within a family than the general population risk, but the patterns of inheritance differ from the classic mendelian autosomal or sex-linked models.
• Most affected children have normal parents. This is true for diseases and for quantitative traits such as intelligence; children with both very superior and extremely low intelligence quotients are born to parents with average intelligence quotients.
• Recurrence risk increases with the number of affected children in a family and with the severity of the defect. More severely affected parents are more likely to produce a more severely affected child.
• Consanguinity slightly increases the risk for having an affected child.
• If the two parents have a different probability of being affected, the parent with the least probability is the one most likely to produce an affected child.
• Genetic counseling for multifactorially or polygenically inherited conditions can be difficult and depends, in large part, on diagnostic clarity and empirical data.
• Although frequently used to describe malformation disorders, multifactorial inheritance and polygenic inheritance have been implicated in such common abnormalities as some forms of cancer and predisposition to coronary artery disease, diabetes, and alcoholism.
• Categorization of polygenic and multifactorially inherited conditions (4).
• Variable traits have a normal distribution in the general population and are measurable or quantitative traits. In these traits, the statistical principle of regression to the mean governs the bell-shaped curve without further extension of the extremes (9).
• Threshold traits are phenotypic traits that do not appear until a critical threshold is met. As such, they are an all-or-none phenomenon with greater likelihood of achieving the threshold within families and with the same environmental impacts. Some of these traits have a sex predilection (likely an environmental effect) (16).
• Complex disorders or traits involve the presence of multiple genes, the environment, and other unknown factors with familial tendencies. They may include obesity, diabetes, hypertension, and some cancers.
• Pharmacogenetics: The effects of medications and their degradation are different in different people; this is genetically based. Enzymes encoded by genes with polymorphic alleles may have different catabolic rates and routes that lead to different reactions to medications.
• Malignant hyperthermia: mutations in the ryanodine receptor protein (16)
• Serum pseudocholinesterase deficiency: demonstrates variations in percentages of enzyme activities
• Adverse reactions to drugs demonstrated on a genetic basis:
 Coumadin
Coumadin
 Isoniazid
Isoniazid
 Corticosteroids
Corticosteroids
• Racial differences in the efficacy of drugs and drug combinations: BiDil, a combination of drugs hydralazine and isosorbide dinitrate for heart failure in African Americans (17)
Table 29-1 Common Congenital Malformations with Multifactorial Inheritance

From Thompson MW, McInnes RR, Willard HF. Thompson & Thompson Genetics in Medicine. 5th ed. Philadelphia: W.B. Saunders, 1991.
MOLECULAR BIOLOGY OF GENETICS
Background
• The gene.
• The genetic material, encoded into packets of information called genes, forms the backbone of the cytogenetically visible chromosome. The estimated 30,000 genes within the human genome, 2 m of DNA, are strung, rather like beads on a string or railroad cars in a train, along the length of the chromosome.
• This linear array of genetic information and the corresponding linear order of the genetic material allow construction of the linear genetic map.
• Genes can be ordered with respect to one another, genetically or physically linked to one another, and physically linked to a particular chromosome (18).
• The genome. Only a small percentage of the genetic information that resides within the human genome can be currently interpreted. About 75% of the DNA is single copy, unique, with only 10% coding for proteins and the remainder being various classes of repetitive DNA (19). The goal of the Human Genome Project is to decipher and understand the human genetic code, promising a more complete understanding of the complex interaction among genes, their expression, and the environment (7).
• The information present within the genetic material of the human is encoded within the polymeric macromolecule DNA.
• Packaged within the double five-carbon sugar backbone are millions of nitrogencontaining bases called pyrimidines (thymine and cytosine) and purines (adenine and guanine).
• The unwinding of the double helix allows precise duplication of the DNA so that daughter cells are given the best possible opportunity of having identical genetic information to the parent cell. However, errors do occur. A replication error in the precise matching of the base sequence of the DNA can result in a base change and therefore a mutation at that site. The mutation may have a significant effect on the daughter cell, or it may have none. Varied alleles at a specific DNA locus are likely prior mutations that have been carried on (2,19).
• It is the precise order of the bases within a length of DNA that defines the expression of the genetic material. The structure of the DNA allows that code to be read and translated into a largely single-stranded transmission molecule, RNA. The RNA molecule, in turn, directs the production of a specific protein of specific amino acid sequence. It is, therefore, the order of the bases within the DNA backbone and the genetic code that define the protein product (http://hyperphysics.phy-astr.gsu.edu/Nave-html/Faithpathh/codelife2.html) (20).
• The code is precise. Even a single base change can have a profound effect on the ultimate protein product and therefore on its activity and expression.
• Mutations occur when chemical or physical agents lead to base modifications; these agents are called mutagens, and their mechanisms of action include
 Deamination of cytosine, adenine, or guanine
Deamination of cytosine, adenine, or guanine
 Methylation of carbon in the nucleic acids
Methylation of carbon in the nucleic acids
 Depurination of DNA by thermal fluctuations in the cell
Depurination of DNA by thermal fluctuations in the cell
 Alkylation of the nucleic acids
Alkylation of the nucleic acids
 Replacement with nucleic acid analogues (e.g., 5-bromodeoxyuridine for thymine)
Replacement with nucleic acid analogues (e.g., 5-bromodeoxyuridine for thymine)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


