 Figure 37–1. Normal male and female human karyotype. (Courtesy of the Colorado Genetics Laboratory.)
Figure 37–1. Normal male and female human karyotype. (Courtesy of the Colorado Genetics Laboratory.)
Cell Division
Cells undergo cycles of growth and division that are controlled according to their needs and functions.
Mitosis is a kind of cell division, occurring in stages, during which DNA replication takes place and two daughter cells, genetically identical to the original parent cells, are formed. This cell division is typical for all somatic cells (cells other than the sperm or egg, which are called germline cells). There are four phases of mitosis: interphase, prophase, metaphase, and anaphase. In interphase, chromosomes are long, thin, and nonvisible. At this time, the genetic material is replicated. In prophase, the chromosomes are more condensed. During metaphase (the phase following DNA replication but preceding cell division), individual chromosomes can be visualized. Each arm consists of two identical parts, called chromatids. Chromatids of the same chromosome are called sister chromatids. In anaphase, the genetic material is separated into two cells.
Meiosis is a kind of cell division during which eggs and sperm are formed; it is a cell division limited to gametes. During meiosis, three unique processes take place:
1. Crossing over of genetic material between two homologous chromosomes (this recombination, or exchange of genetic material increases the viability of human beings).
2. Random assortment of maternally and paternally derived homologous chromosomes into the daughter cells.
3. Two cell divisions, the first of which is a reduction division—that is, separation between the homologous chromosomes. The second meiotic division is like mitosis, separating two sister chromatids into two daughter cells.
Chromosome Preparation & Analysis
Chromosome structure is visible only during mitosis, most often achieved in the laboratory by stimulating a blood lymphocyte culture with a mitogen for 3 days. Other tissues used for this purpose include skin, products of conception, cartilage, and bone marrow. Chorionic villi or amniocytes are used for prenatal diagnosis. Spontaneously dividing cells without a mitogen are present in bone marrow, and historically, bone marrow biopsy was done when immediate identification of a patient’s chromosome constitution was necessary for appropriate management (eg, to rule out trisomy 13 in a newborn with a complex congenital heart disease). However, this invasive test has been replaced by the availability of the FISH technique (see the following discussion).
Cells processed for routine chromosome analysis are stained on glass slides to yield a light-and-dark band pattern across the arms of the chromosomes (see Figure 37–1). This band pattern is characteristic and reproducible for each chromosome, allowing the chromosomes to be identified. Using different staining techniques, different banding patterns result: G, Q, and R banding. The most commonly used is G banding. The layout of chromosomes on a sheet of paper in a predetermined order is called a karyotype. High-resolution chromosome analysis is the study of more elongated chromosomes in prometaphase. Although the bands can be visualized in greater detail, subtle chromosomal rearrangements less than 5 million base pair (5 Mb) can still be missed.
Fluorescence in situ hybridization (FISH) is a powerful technique that labels a known chromosome sequence with DNA probes attached to fluorescent dyes, thus enabling visualization of specific regions of chromosomes by fluorescent microscopy. There are many different kinds of probes, including paint probes (a mixture of sequences throughout one chromosome), sequence-specific probes, centromere probes, and telomere probes. A cocktail of differently colored probes, one color for each chromosome, called multicolor FISH, or M-FISH, can detect complex rearrangements between chromosomes. FISH can detect submicroscopic structural rearrangements undetectable by classic cytogenetic techniques and can identify marker chromosomes. (For pictures of FISH studies, go to http://www.kumc.edu/gec/prof/cytogene.html.)
Interphase FISH allows noncultured cells (lymphocytes, amniocytes) to be rapidly screened for numerical abnormalities such as trisomy 13, 18, or 21, and sex chromosome anomalies. However, because of the possible background or contamination of the signal, the abnormality must be confirmed by conventional chromosome analysis in aneuploidy cases. Six hundred–cell FISH can also be used to ascertain mosaicism.
Chromosomal Microarray Analysis
Advances in computer technology and bioinformatics have led to the development of new genetic testing using comparative genomic hybridization with microarray technique. This technique allows detection of very small genetic imbalances anywhere in the genome. Its usefulness has been well documented in cancer research and more recently in assessing small chromosomal rearrangements. In particular, it has been used to detect interstitial and subtelomeric submicroscopic imbalances, to characterize their size at the molecular level, and to define the breakpoints of translocations. Clinically available arrays include (1) 0.5- to 1-Mb bacterial artificial chromosome arrays that can pick up rearrangements greater than 0.5 Mb, (2) oligonucleotide arrays using special probes that can pick up changes as small as 3 Kb, and (3) single nucleotide polymorphism (SNP) arrays, which are used more widely in research settings. Although this powerful new technology can identify extremely subtle DNA rearrangements and changes, many human polymorphisms, including small deletions and duplications, are not totally understood. Therefore, special caution and parental studies are often required in interpreting the results.
Stankiewicz P, Beaudet AL: Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr Opin Genet Dev 2007 Jun;17(3):182–192 [Epub 2007 Apr 30] [Review] [PMID: 17467974].
Van den Veyver IB et al: Clinical use of array comparative genomic hybridization (aCGH) for prenatal diagnosis in 300 cases. Prenat Diagn 2009 Jan;29(1):29–39 [PMID: 19012303].
Chromosome Nomenclature
Visible under the microscope is a constriction site on the chromosome called the centromere, which separates the chromosome into two arms: p, for petite, refers to the short arm, and q, the letter following p, refers to the long arm. Each arm is further subdivided into numbered bands visible using different staining techniques. Centromeres are positioned at different sites on different chromosomes and are used to differentiate the chromosome structures seen during mitosis as metacentric (p arm and q arm of almost equal size), submetacentric (p arm shorter than q arm), and acrocentric (almost no p arm). The use of named chromosome arms and bands provides a universal method of chromosome description. Common symbols include del (deletion), dup (duplication), inv (inversion), ish (in situ hybridization), i (isochromosome), pat (paternal origin), mat (maternal origin), and r (ring chromosome). These terms are further defined in the section Chromosomal Abnormalities.
Chromosomal Abnormalities
There are two types of chromosomal anomalies: numerical and structural.
A. Abnormalities of Chromosomal Number
When a human cell has 23 chromosomes, such as human ova or sperm, it is in the haploid state (n). After conception, in cells other than the reproductive cells, 46 chromosomes are present in the diploid state (2n). Any number that is an exact multiple of the haploid number—for example, 46(2n), 69(3n), or 92(4n)—is referred to as euploid. Polyploid cells are those that contain any number other than the usual diploid number of chromosomes. Polyploid conceptions are usually not viable except in a “mosaic state,” with the presence of more than one cell line in the body (see later text for details).
Cells deviating from the multiple of the haploid number are called aneuploid, meaning not euploid, indicating an abnormal number of chromosomes. Trisomy, an example of aneuploidy, is the presence of three of a particular chromosome rather than two. It results from unequal division, called nondisjunction, of chromosomes into daughter cells. Trisomies are the most common numerical chromosomal anomalies found in humans (eg, trisomy 21 [Down syndrome], trisomy 18, and trisomy 13). Monosomies, the presence of only one member of a chromosome pair, may be complete or partial. Complete monosomies may result from nondisjunction or anaphase lag. All complete autosomal monosomies appear to be lethal early in development and only survive in mosaic forms. Sex chromosome monosomy, however, can be viable.
B. Abnormalities of Chromosomal Structure
Many different types of structural chromosomal anomalies exist. Figure 37–2 displays the formal nomenclature as well as the ideogram demonstrating chromosomal anomalies. In clinical context, the sign (+) or (−) preceding the chromosome number indicates increased or decreased number, respectively, of that particular whole chromosome in a cell. For example, 47, XY+21 designates a male with three copies of chromosome 21. The sign (+) or (−) after the chromosome number signifies extra material or missing material, respectively, on one of the arms of the chromosome. For example, 46, XX, 8q− denotes a deletion on the long arm of chromosome 8. Detailed nomenclature, such as 8q11, is required to further demonstrate a specific missing region so that genetic counseling can be provided.
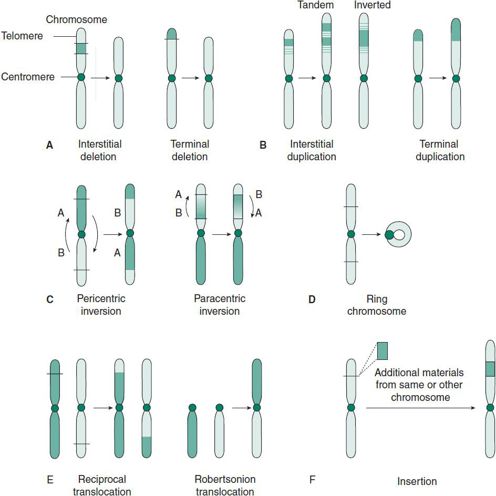
 Figure 37–2. Examples of structural chromosomal abnormalities: deletion, duplication, inversion, ring chromosome, translocation, and insertion.
Figure 37–2. Examples of structural chromosomal abnormalities: deletion, duplication, inversion, ring chromosome, translocation, and insertion.
1. Deletion (del) (see Figure 37–2A)—This refers to an absence of normal chromosomal material. It may be terminal (at the end of a chromosome) or interstitial (within a chromosome). The missing part is described using the code “del,” followed by the number of the chromosome involved in parentheses, and a description of the missing region of that chromosome, also in parentheses, for example, 46, XX, del(1) (p36.3). This chromosome nomenclature describes the loss of genetic material from band 36.3 of the short arm of chromosome 1, which results in 1p36.3 deletion syndrome. Some more common deletions result in clinically recognizable conditions associated with intellectual disabilities and characteristic facial features. (See descriptions of common genetic disorders caused by chromosomal deletions later in the chapter.)
2. Duplication (dup) (see Figure 37–2B)—An extra copy of a chromosomal segment can be tandem (genetic material present in the original direction) or inverted (genetic material present in the opposite direction). A well-described duplication of chromosome 22q11 causes Cat eye syndrome, resulting in iris coloboma and anal or ear anomalies.
3. Inversion (inv) (see Figure 37–2C)—In this aberration, a rearranged section of a chromosome is inverted. It can be paracentric (not involving the centromere) or pericentric (involving the centromere).
4. Ring chromosome (r) (see Figure 37–2D)—Deletion of the normal telomeres (and possibly other subtelomeric sequences) leads to subsequent fusion of both ends to form a circular chromosome. Ring chromosomal anomalies often cause growth retardation and intellectual disability.
5. Translocation (trans) (see Figure 37–2E)—This interchromosomal rearrangement of genetic material may be balanced (the cell has a normal content of genetic material arranged in a structurally abnormal way) or unbalanced (the cell has gained or lost genetic material as a result of chromosomal interchange). Balanced translocations may further be described as reciprocal, the exchange of genetic material between two nonhomologous chromosomes, or Robertsonian, the fusion of two acrocentric chromosomes.
6. Insertion (ins) (see Figure 37–2F)—Breakage within a chromosome at two points and incorporation of another piece of chromosomal material is called insertion. This requires three breakpoints and may occur between two chromosomes or within the same chromosome. The clinical presentation or phenotype depends on the origin of the inserted materials.
C. Sex Chromosomal Anomalies
Abnormalities involving sex chromosomes, including aneuploidy and mosaicism, are relatively common in the general population. The most common sex chromosome anomalies include 45,X (Turner syndrome), 47,XXX,47,XXY (Klinefelter syndrome), 47,XYY, and different mosaic states. (See later text for clinical discussion.)
D. Mosaicism
Mosaicism is the presence of two or more different chromosome constitutions in different cells of the same individual. For example, a patient may have some cells with 47 chromosomes and others with 46 chromosomes (46,XX/47,XX,+21 indicates mosaicism for trisomy 21; similarly, 45,X/46,XX/47,XXX indicates mosaicism for a monosomy and a trisomy X). Mosaicism should be suspected if clinical symptoms are milder than expected in a nonmosaic patient with the same chromosomal abnormality, or if the patient’s skin shows unusual pigmentation. The prognosis is frequently better for a patient with mosaicism than for one with a corresponding chromosomal abnormality without mosaicism. In general, the smaller the proportion of the abnormal cell line, the better the prognosis. In the same patient, however, the proportion of normal and abnormal cells in various tissues, such as skin, brain, internal organs, and peripheral blood, may be significantly different. Therefore, the prognosis for a patient with chromosomal mosaicism can seldom be assessed reliably based on the karyotype in peripheral blood alone.
E. Uniparental Disomy
Under normal circumstances, one member of each homologous pair of chromosomes is of maternal origin from the egg and the other is of paternal origin from the sperm (Figure 37–3A). In uniparental disomy (UPD), both copies of a particular chromosome pair originate from the same parent. If UPD is caused by an error in the first meiotic division, both homologous chromosomes of that parent will be present in the gamete—a phenomenon called heterodisomy (Figure 37–3B). If the disomy is caused by an error in the second meiotic division, two copies of the same chromosome will be present through the mechanism of rescue, duplication, and complementation (Figure 37–3C through 37–3E)—a phenomenon called isodisomy. Isodisomy may also occur as a postfertilization error (Figure 37–3F).
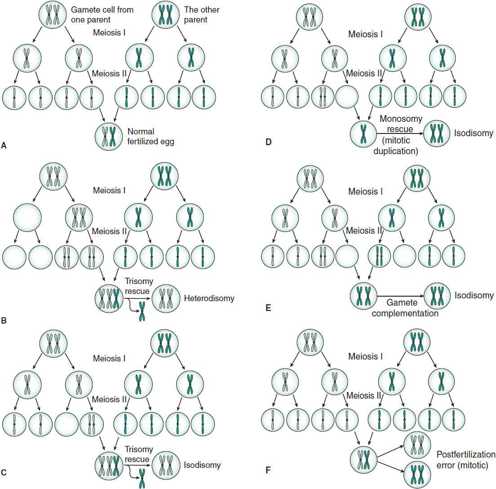
 Figure 37–3. The assortment of homologous chromosomes during normal gametogenesis and uniparental disomy. A: Fertilization of normal gametes. B: Heterodisomy by trisomy rescue. C: Isodisomy by trisomy rescue. D: Isodisomy by monosomy rescue (mitotic duplication). E: Gamete complementation. F: Postfertilization error.
Figure 37–3. The assortment of homologous chromosomes during normal gametogenesis and uniparental disomy. A: Fertilization of normal gametes. B: Heterodisomy by trisomy rescue. C: Isodisomy by trisomy rescue. D: Isodisomy by monosomy rescue (mitotic duplication). E: Gamete complementation. F: Postfertilization error.
A chromosomal analysis would not reveal an abnormality, but DNA analysis would reveal that the child inherited two copies of DNA of a particular chromosome from one parent without the contribution from the other parent. Possible mechanisms for the adverse effects of UPD include homozygosity for deleterious recessive genes and the consequences of imprinting (see discussion in the Imprinting section, later). It is suspected that UPD of some chromosomes is lethal.
UPD has been documented for certain human chromosomes, including chromosomes 7, 11, 15, and X, and has been found in patients with Prader-Willi, Angelman, and Beckwith-Wiedemann syndromes (BWS). In addition, cystic fibrosis with only one carrier parent (caused by maternal isodisomy) has been reported. UPD may cause severe prenatal and postnatal growth retardation.
F. Contiguous Gene Syndromes
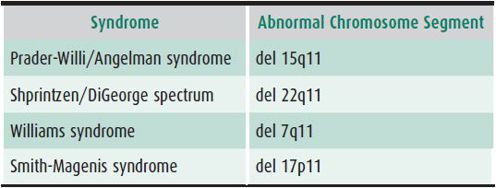
Contiguous gene syndromes result when a deletion causes the loss of genes adjacent to each other on a chromosome. Although many genes may be missing, the deletion may still be too small to be detected by routine karyotype. Therefore, contiguous gene syndromes are sometimes called “microdeletion syndromes.” The genes involved in these syndromes are related only through their linear placement on the same chromosome segments and may not influence each other’s functions directly. Table 37–1 lists examples of some currently known contiguous gene syndromes and their associated chromosomal abnormalities. These deletions may be familial (passed on by a parent) or may occur de novo. The deletions may be diagnosed by high-resolution chromosome analysis in some affected individuals, or may be submicroscopic and detectable only with FISH or DNA analysis.
Table 37–1. Examples of common contiguous gene syndromes.
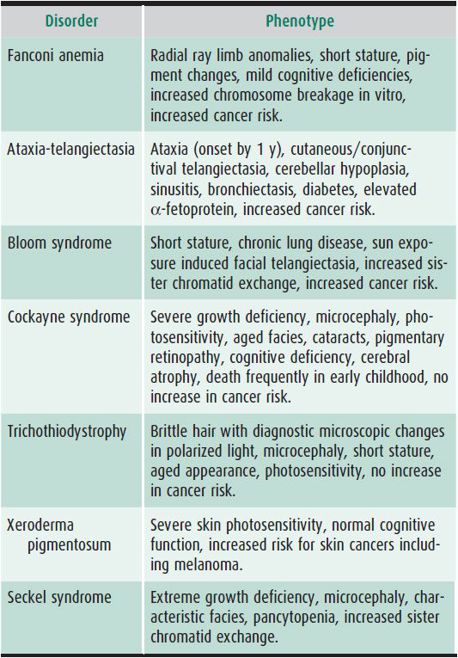
G. Chromosome Fragility
Disorders of DNA repair are associated with chromosomal breakage and death of somatic cells. Most are autosomal recessive. Phenotypes vary considerably (Table 37–2). As a group these disorders typically affect growth and CNS development. They show increased toxicity to mutagen exposures in vitro. Photosensitivity, increased cancer risks, and premature aging are prevalent. Treatment is largely supportive and focuses on surveillance for complications but in at least one disorder, Fanconi anemia, bone marrow transplant can be beneficial. See www.genereviews.org for excellent reviews of these disorders.
Table 37–2. DNA repair disorders.
H. Chromosomal Abnormalities in Cancer
Numerical and structural chromosomal abnormalities are often identified in hematopoietic and solid-tumor neoplasms in individuals with otherwise normal chromosomes. These cytogenetic abnormalities have been categorized as primary and secondary. In primary abnormalities, their presence is necessary for initiation of the cancer; an example is 13q− in retinoblastoma. Secondary abnormalities appear de novo in somatic cells only after the cancer has developed, for example, Philadelphia chromosome, t(9;22)(q34;q11), in acute and chronic myeloid leukemia. Primary and secondary chromosomal abnormalities are specific for particular neoplasms and can be used for diagnosis or prognosis. For example, the presence of the Philadelphia chromosome is a good prognostic sign in chronic myelogenous leukemia and indicates a poor prognosis in acute lymphoblastic leukemia. The sites of chromosome breaks coincide with the known loci of oncogenes and antioncogenes.
MOLECULAR GENETICS
Advances in molecular biology have revolutionized human genetics, as they allow for the localization, isolation, and characterization of genes that encode protein sequences. As the Human Genome Project has moved into the postcloning era, the function of gene products and their interaction with one another has become the main theme of molecular genetics. Molecular genetics can help explain the complex underlying biology involved in many human diseases.
Molecular diagnosis can be achieved using the following technology: Southern blot analysis is the molecular genetic technique used to look for changes in genomic DNA. A similar technique, called Northern blot analysis, is used to look for RNA abnormalities. Western blot analysis is used to look for protein changes. The polymerase chain reaction (PCR) replicates fragments of DNA between predetermined primers so that sufficient DNA is obtained for characterization or sequencing in the space of a few hours. Quantitative fluorescent PCR combines PCR amplification with fluorescent DNA probes to provide real-time replication and rapid determination of gene copy number and dosage effects. DNA sequencing is the process of determining the nucleotide order of a given DNA fragment. A new generation of sequencing technologies has provided unprecedented opportunities for high-throughput functional genomic research. To date, these technologies have been applied in a variety of contexts, including whole-genome sequencing which can be performed in 1 week; however, the interpretation of the sequencing requires more bioinformatics information. National Institutes of Health (NIH) predicted the eventual cost for whole genome can be reduced to $1000.
ten Bosch JR, Grody WW: Keeping up with the next generation: massively parallel sequencing in clinical diagnostics. J Mol Diagn 2008 Nov;10(6):484–492 [PMID: 18832462].
Molecular Biology in Clinical Genetics & Genetic Diagnosis
Genetic diagnosis can be performed by direct detection of a mutant gene or by indirect methods. Direct detection is possible only when the gene causing the disease and the nature of the mutation are known. The advantage of a diagnostic study using the direct detection of a mutant gene is that it requires the affected individual only and need not involve the testing of other family members. The methods of direct DNA diagnosis include restriction analysis, direct sequencing with assistance of PCR, heteroduplex assay, and protein truncation assay. The molecular mechanisms causing human diseases include point mutations, deletions, and insertions, and the unstable expansion of trinucleotide repeats, which leads to genetic anticipation. Some disorders that may be diagnosed via direct DNA mutational analysis include Duchenne muscular dystrophy, hemophilia, cystic fibrosis, and Fragile X syndrome.
Indirect detection of abnormal genes is used when the gene is known but there is extensive heterogeneity of the molecular defect between families, or when the gene responsible for a disease is unknown but its chromosome location is known. One form of indirect analysis is the linkage method. Linkage traces the inheritance of the abnormal gene between members in a kindred. This method requires that the affected individual be studied, as well as parents and other relatives, both affected and unaffected. Linkage analysis is performed by using markers such as a restriction fragment length polymorphisms. Microsatellite polymorphisms are being used in sibling research studies to identify the multiple genes that contribute to polygenic traits such as diabetes and obesity. They are also used increasingly to identify gene changes in tumors.
Neurofibromatosis is an example of a disorder in which both the direct and indirect assay may be used. An estimated 90%–95% of patients with neurofibromatosis type 1 have a mutation or deletion that can be identified using a direct assay of the neurofibromin gene (NF1). The other cases must rely on indirect methods such as linkage analysis for prenatal diagnosis.
Molecular Biology in Prevention & Treatment of Human Diseases
Molecular diagnosis can prevent genetic disease by detection of mutation and permitting prenatal diagnosis. As diseases often present in spectrums and clinical features among disorders can overlap, molecular testing is useful to confirm a diagnosis. Family studies can also clarify the mode of inheritance, thus allowing more accurate determination of recurrence risks and appropriate options. For example, differentiation of gonadal mosaicism from decreased penetrance of a dominant gene has important implications for genetic counseling. In the past, the diagnosis of a genetic disease characterized by late onset of symptoms (eg, Huntington disease) could not be made prior to the appearance of clinical symptoms. In some inborn errors of metabolism, diagnostic tests (eg, measurement of enzyme activities) could be conducted only on inaccessible tissues. Gene identification (mutation analysis) techniques can enormously enhance the ability to diagnose both symptomatic and presymptomatic individuals, heterozygous carriers of gene mutations, and affected fetuses. However, presymptomatic DNA testing is associated with psychological, ethical, and legal implications and therefore should be used only with informed consent. Formal genetic counseling is indicated to best interpret the results of molecular testing.
A normal gene introduced into an individual affected with a serious inherited disorder during embryonic life (germline therapy) in principle has the potential to be transmitted to future generations, whereas its introduction into somatic cells (somatic therapy) affects only the recipient. Experimental gene therapy by bone marrow transplantation is being tried for adenosine deaminase deficiency. Recombinant enzyme replacement has been successfully applied in treating the nonneurologic form of Gaucher disease, Fabry disease, Pompe disease, mucopolysaccharidosis types I and II, and some types of lysosomal storage disease.
Proteomics is the large-scale study of proteins, particularly their structures and functions. The term “proteomics” was first coined in 1997 as an analogy to genomics, the study of the genes. “Proteome” means a blend of “protein” and “genome.” Understanding the proteome, the structure and function of each protein and the complexities of protein-protein interactions will be critical for developing effective diagnostic techniques and disease treatments. One of the most promising roles of proteomics has been the identification of potential new drugs for the treatment of disease. This relies on genome and proteome information to identify proteins associated with a disease, which computer software can then use as targets for new drugs. For example, in Alzheimer disease, elevations in beta secretase create amyloid/beta-protein, which causes plaque to build up in the patient’s brain, which is thought to play a role in dementia. Targeting this enzyme decreases the amyloid/beta-protein and so slows the progression of the disease.
Pharmacogenomics is a new field offering enormous promise for predicting drug response in patients. For example, by DNA analysis of two specific genes, CYP2C9 and VKORC1, it is now possible to predict response to warfarin anticoagulation therapy and to individualize the dose, saving the patient multiple blood tests and dosage adjustments. It is also possible to predict which patients would be at risk for hearing loss after receiving aminoglycoside treatment, based on mutations in the mitochondrial 12S rRNA gene.
Personalized medicine is an advancing field of medicine that offers increased precision and effectiveness than traditional medicine. A patient’s genomic information offers insight into the individual aspects of one’s medical management. The goal is to optimize care and overall outcomes, one example being the aforementioned area of pharmacogenomics and potential therapeutic responses.
Genetic testing allows practitioners to test patients for a wide variety of genetic conditions. Advances in this area of medicine have included the advent of parents requesting testing for adult onset disease, carrier status, and disease susceptibility in their children. There are significant ethical and legal issues surrounding this topic. The American College of Medical Genetics and Genomics and American Society of Human Genetics formed a consensus statement on the topic that educates families and healthcare providers on the potential negative impacts of such testing. In the face of whole exome or genome sequencing, single-gene analysis, and microarray analysis, carrier status for conditions may be revealed and this requires detailed genetic counseling. The decision-making capacity of the minor should also be taken into account where applicable.
Friedman Ross L et al: Technical report: ethical and policy issues in genetic testing and screening of children. Genet Med 2013;15(3):234–245 [PMID: 23429433].
Garcia DA et al: Estimation of the warfarin dose with clinical and pharmacogenetic data. NEJM 2009;360:753–764 [PMID: 19228618].
Ma Q et al: Pharmacogenetics, pharmacogenomics, and individualized medicine. Pharmacol Rev 2011 Jun;63(2):437–459 [Epub 2011 Mar 24] [PMID: 21436344].
PRINCIPLES OF INHERITED HUMAN
MENDELIAN INHERITANCE
Traditionally, autosomal single-gene disorders follow the principles explained by Mendel’s observations. To summarize, the inheritance of genetic traits through generations relies on segregation and independent assortment. Segregation is the process through which gene pairs are separated during gamete formation. Each gamete receives only one copy of each gene (allele). Independent assortment refers to the idea that the segregation of different alleles occurs independently.
Victor McKusick’s catalog, Mendelian Inheritance in Man, lists more than 10,000 entries in which the mode of inheritance is presumed to be autosomal dominant, autosomal recessive, X-linked dominant, X-linked recessive, and Y-linked. Single genes at specific loci on one or a pair of chromosomes cause these disorders. An understanding of inheritance terminology is helpful in approaching mendelian disorders. Analysis of the pedigree and the pattern of transmission in the family, identification of a specific condition, and knowledge of that condition’s mode of inheritance usually allow for explanation of the inheritance pattern.
Terminology
The following terms are important in understanding heredity patterns.
1. Dominant and recessive—As defined by Mendel, concepts for dominant and recessive refer to the phenotypic expression of alleles and are not intrinsic characteristics of gene loci. Therefore, it is inappropriate to discuss “a dominant locus.”
2. Genotype—Genotype means the genetic status, that is, the alleles an individual carries.
3. Phenotype—Phenotype is the expression of an individual’s genotype including appearance, physical features, organ structure, and biochemical and physiologic nature. It may be modified by environment.
4. Pleiotropy—Pleiotropy refers to the phenomenon whereby a single mutant allele can have widespread effects or expression in different tissues or organ systems. In other words, an allele may produce more than one effect on the phenotype. For example, Marfan syndrome has manifestations in different organ systems (skeletal, cardiac, ophthalmologic, etc) due to a single mutation within the fibrillin gene.
5. Penetrance—Penetrance refers to the proportion of individuals with a particular genotype that express the same phenotype. Penetrance is a proportion that ranges between 0 and 1 (or 0 and 100%). When 100% of mutant individuals express the phenotype, penetrance is complete. If some mutant individuals do not express the phenotype, penetrance is said to be incomplete, or reduced. Dominant conditions with incomplete penetrance, therefore, are characterized by “skipped” generations with unaffected, obligate gene carriers.
6. Expressivity—Expressivity refers to the variability in degree of phenotypic expression (severity) seen in different individuals with the same mutant genotype. Expressivity may be extremely variable or fairly consistent, both within and between families. Intrafamilial variability of expression may be due to factors such as epistasis, environment, genetic anticipation, presence of phenocopies, mosaicism, and chance (stochastic factors). Interfamilial variability of expression may be due to the previously mentioned factors, but may also be due to allelic or locus genetic heterogeneity.
7. Genetic heterogeneity—Several different genetic mutations may produce phenotypes that are identical or similar enough to have been traditionally considered as one diagnosis. “Anemia” or “mental retardation” are examples of this. There are two types of genetic heterogeneity, locus heterogeneity and allelic heterogeneity.
A. LOCUS HETEROGENEITY—Locus heterogeneity describes a phenotype caused by mutations at more than one genetic locus; that is, mutations at different loci cause the same phenotype or a group of phenotypes that appear similar enough to have been previously classified as a single disease, clinical “entity,” or diagnostic spectrum. An example would be Sanfilippo syndrome (mucopolysaccharidosis types IIIA, B, C, and D), in which the same phenotype is produced by four different enzyme deficiencies.
B. ALLELIC HETEROGENEITY—A phenotype causing different mutations at a single-gene locus. As an example, cystic fibrosis may be caused by many different genetic changes, such as homozygosity for the common Δ F508 mutation, or ΔF508 and an R117H mutation. The latter example represents compound heterozygosity.
8. Phenotypic heterogeneity or “clinical heterogeneity”—This term describes the situation in which more than one phenotype is caused by different allelic mutations at a single locus. For example, different mutations in the FGFR2 gene can cause different craniosynostosis disorders, including Crouzon syndrome, Jackson-Weiss syndrome, Pfeiffer syndrome, and Apert syndrome. These syndromes are clinically distinguishable and are due to the presence of a variety of genetic mutations within single genes.
9. Homozygous—A cell or organism that has identical alleles at a particular locus is said to be homozygous. For example, a cystic fibrosis patient with a ΔF508 mutation on both alleles would be called homozygous for that mutation.
10. Heterozygous—A cell or organism that has nonidentical alleles at a genetic locus is said to be heterozygous. In autosomal dominant conditions, a mutation of only one copy of the gene pair is all that is necessary to result in a disease state. However, an individual who is heterozygous for a recessive disorder will not manifest symptoms (see the next section).
11. Karyotype—A profile of an organism’s chromosomes that is sorted according to size, shape, and number. It is available for analysis in a number of sample types (white blood cells, fibroblasts, etc). It is able to detect structural rearrangements such as inversions, positional insertions, and translocations. Imbalances below 5 million base pairs are difficult to detect.
12. Chromosomal microarray—Method of cytogenetic analysis via several platforms: BAC, oligonucleotide, and SNP. Patient DNA is hybridized with control DNA and each is labeled with a different fluorescent dye. Data is plotted on a log2 scale, as in the case of oligonucletide arrays, and reviewed for numerical imbalances. Microarray is limited in the detection of balanced rearrangements and the structural nature of an imbalance.
13. Next-generation sequence analysis—Genetic analysis via breakage of the genome into fragments, attaching those segments of DNA to special adapters, or passage through specialized channels where the sequence is determined. Millions of segments are analyzed simultaneously, lending the term “high throughput” to this approach. Regions are repeatedly analyzed and compared with a reference human genome.
14. Whole exome sequencing—Determination of the sequence of an individual’s exome, the coding sequence of the human genome. Exome represents only about 1% of the genome.
15. Whole genome sequencing—Determination of the sequence of the entire human genome
Bick D, Dimmock D: Whole exome and whole genome sequencing. Curr Opin Pediatr 2011 Dec;23(6):594–600 [PMID: 21881504].
Online Mendelian Inheritance in Man: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM.
Hereditary Patterns
A. Autosomal Dominant Inheritance
Autosomal dominant inheritance has the following characteristics:
1. If a parent is affected, the risk for each offspring of inheriting the abnormal dominant gene is 50%, or 1:2. This is true whether the gene is penetrant or not in the parent.
2. Affected individuals in the same family may experience variable expressivity.
3. Nonpenetrance is common, and the penetrance rate varies for each dominantly inherited condition.
4. Both males and females can pass on the abnormal gene to children of either sex, although the manifestations may vary according to sex. For example, pattern baldness is a dominant trait but affects only males. In this case, the trait is said to be sex-limited.
5. Dominant inheritance is typically said to be vertical, that is, the condition passes from one generation to the next in a vertical fashion (Figure 37–4).
6. In some cases, the patient appears to be the first affected individual in the family. This spontaneous appearance is often caused by a new mutation. The mutation rate increases with advancing paternal age (particularly after age 40 years).
7. Explanations for a negative family history include:
A. Nonpaternity.
B. Decreased penetrance or mild manifestations in one of the parents.
C. Germline mosaicism (ie, mosaicism in the germ cell line of either parent). Germline mosaicism may mimic autosomal recessive inheritance, because it leads to situations in which two children of completely normal parents are affected with a genetic disorder. Recurrence risks are in the range of 1%–7%.
D. The abnormality present in the patient may be a phenocopy, or it may be a similar but genetically different abnormality with a different mode of inheritance.
8. As a general rule, dominant traits are more often related to structural abnormalities of a protein.
9. If an abnormality represents a new mutation of a dominant trait, the parents of the affected individual run a low risk during subsequent pregnancies. The risk for an affected sibling is still slightly increased over the general population, because of the possibility of germline mosaicism.
10. Prevention options available for future pregnancies include prenatal diagnosis, artificial insemination, and germ cell donation.
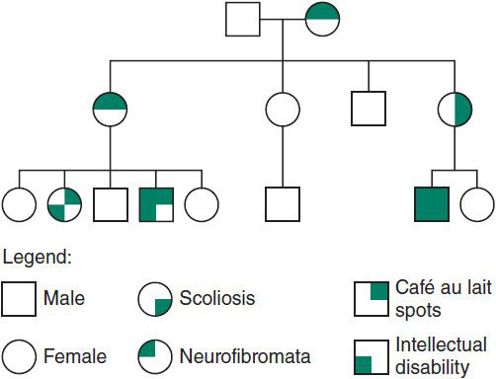
 Figure 37–4. Autosomal dominant inheritance. Variable expressivity in Neurofibromatosis type 1.
Figure 37–4. Autosomal dominant inheritance. Variable expressivity in Neurofibromatosis type 1.
B. Autosomal Recessive Inheritance
Autosomal recessive inheritance also has some distinctive characteristics:
1. The recurrence risk for parents of an affected child is 25%, or 1:4 for each pregnancy. The gene carrier frequency in the general population can be used to assess the risk of having an affected child with a new partner, for unaffected siblings, and for the affected individuals themselves.
2. There is less variability among affected persons. Parents are carriers and are clinically normal. (There are, however, exceptions to this rule. For example, carriers of sickle cell trait may become symptomatic if they become hypoxic.)
3. Males and females are affected equally.
4. Inheritance is horizontal; siblings may be affected (Figure 37–5).
5. The family history is usually negative, with the exception of siblings. However, in common conditions such as cystic fibrosis, a second- or third-degree relative may be affected.
6. Recessive conditions are frequently associated with enzyme defects.
7. In rare instances, a child with a recessive disorder and a normal karyotype may have inherited both copies of the abnormal gene from one parent and none from the other. This UPD was first described in a girl with cystic fibrosis and growth retardation.
8. Options available for future pregnancies include prenatal diagnosis, adoption, artificial insemination, and egg or sperm donation.
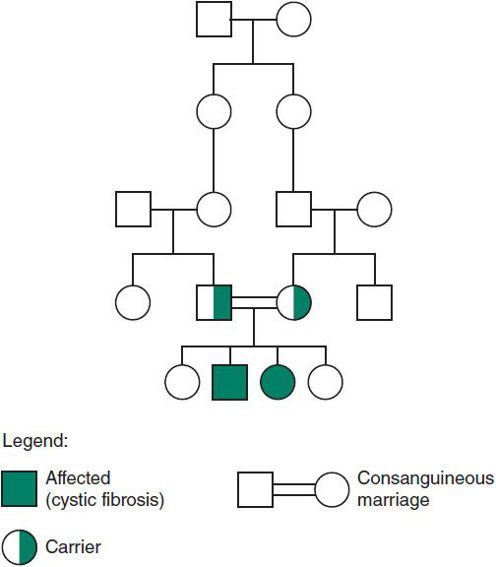
 Figure 37–5. Autosomal recessive inheritance: cystic fibrosis.
Figure 37–5. Autosomal recessive inheritance: cystic fibrosis.
C. X-Linked Inheritance
When a gene for a specific disorder is on the X chromosome, the condition is said to be X-linked, or sex-linked. Females may be either homozygous or heterozygous, because they have two X chromosomes. Males, by contrast, have only one X, and a male is said to be hemizygous for any gene on his X chromosome. The severity of any disorder is greater in males than in females (within a specific family). According to the Lyon hypothesis, because one of the two X chromosomes in each cell is inactivated, and this inactivation is random, the clinical picture in females depends on the percentage of mutant versus normal alleles inactivated. The X chromosome is not inactivated until about 14 days of gestation, and parts of the short arm remain active throughout life.
1. X-linked recessive inheritance—The following features are characteristic of X-linked recessive inheritance:
1. Males are affected, and heterozygous females are either normal or have mild manifestations.
2. Inheritance is diagonal through the maternal side of the family (Figure 37–6A).
3. A female carrier has a 50% chance that each daughter will be a carrier and a 50% chance that each son will be affected.
4. All of the daughters of an affected male are carriers, and none of his sons are affected.
5. The mutation rate is high in some X-linked disorders, particularly when the affected male dies or is so incapacitated by the disorder that reproduction is unlikely. In such instances, the mutation is thought to occur as a new mutation in the affected male, and in the mother, each one-third of the time and to be present in earlier generations one-third of the time. For this reason, genetic counseling may be difficult in families with an isolated case.
6. On rare occasions, a female may be fully affected. Several possible mechanisms may account for a fully affected female: (a) unfavorable lyonization; (b) 45,X karyotype; (c) homozygosity for the abnormal gene; (d) an X-autosome translocation, or other structural abnormality of one X chromosome, in which the X chromosome of normal structure is preferentially inactivated; (e) UPD; and (f) nonrandom inactivation, which may be controlled by an autosomal gene.
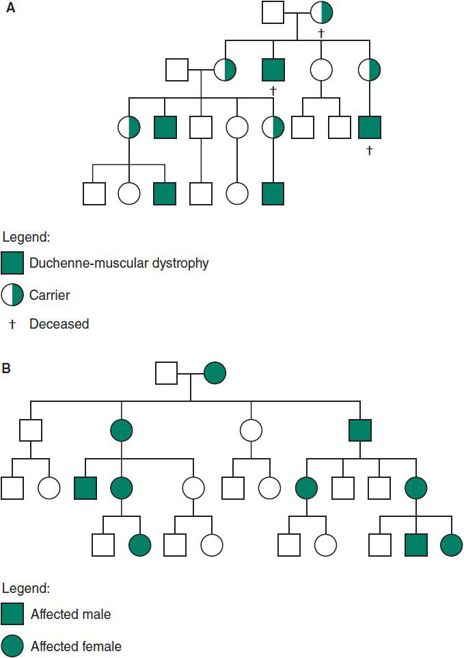
 Figure 37–6. A: X-linked recessive inheritance. B: X-linked dominant inheritance.
Figure 37–6. A: X-linked recessive inheritance. B: X-linked dominant inheritance.
2. X-linked dominant inheritance—The X-linked dominant inheritance pattern is much less common than the X-linked recessive type. Examples include incontinentia pigmenti and hypophosphatemic or vitamin D–resistant rickets. The following features are characteristic of X-linked dominant inheritance:
1. The heterozygous female is symptomatic, and the disease is twice as common in females because they have two X chromosomes that can have the mutation.
2. Clinical manifestations are more variable in females than in males.
3. The risk for the offspring of heterozygous females to be affected is 50% regardless of sex.
4. All of the daughters but none of the sons of affected males will have the disorder (Figure 37–6B).
5. Although a homozygous female is possible (particularly in an inbred population), she would be severely involved. All of her children would also be affected but more mildly.
6. Some disorders (eg, incontinentia pigmenti) are lethal in males (and in homozygous females). Affected women have twice as many daughters as sons and an increased incidence of miscarriages, because affected males will be spontaneously aborted. A 47,XXY karyotype has allowed affected males to survive.
D. Y-Linked Inheritance
In Y-linked inheritance, also known as “holandric” inheritance, a disorder is caused by genes located on the Y chromosome. These conditions are relatively rare with only about 40 entries listed in McKusick’s catalog. Male-to-male transmission is seen in this category, with all sons of affected males being affected and no daughters or females being affected.
MULTIFACTORIAL INHERITANCE
Many common attributes, such as height, are familial, and are the result of the actions of multiple rather than single genes. Inheritance of these traits is described as polygenic or multifactorial. The latter term recognizes that environmental factors such as diet also contribute to these traits. Geneticists are now finding that multiple genes are often expressed in hierarchies, in which the action of a small number of genes, two or three, explains much of the variation observed within affected populations.
Studies of twins have proven useful in determining the relative importance of genetic versus environmental factors in the expression of polygenic traits. If genetic factors are of little or no importance, then the concordance between monozygotic and dizygotic twins should be the same. (Dizygotic twins are no more genetically similar to each other than to other siblings.) If an abnormality is completely genetic, the concordance between identical twins should be 100%. In polygenic conditions, the concordance rate for identical twins is usually higher than that seen in dizygotic twins but is still not 100%, indicating that both genetic and environmental factors are playing a role.
Many disorders and congenital abnormalities that are clearly familial but do not segregate as mendelian traits (eg, autosomal dominant, recessive) show polygenic inheritance. For the most part, these conditions become manifest when thresholds of additive gene actions or contributing environmental factors are exceeded. Many common disorders ranging from hypertension, stroke, and thrombophlebitis to behavioral traits such as alcoholism demonstrate multifactorial (polygenic) inheritance. Some common birth defects, including isolated congenital heart disease, cleft lip and palate, and neural tube defects, also demonstrate polygenic inheritance. Neural tube defects provide a good model illustrating how identification of both environmental and genetic contributions to multifactorial traits can lead to preventive measures.
Polygenic or multifactorial inheritance has several distinctive characteristics:
1. The risk for relatives of affected persons is increased. The risk is higher for first-degree relatives (those who have 50% of their genes in common) and lower for more distant relations, although the risk for the latter is higher than for the general population (Table 37–3).
Table 37–3. Empiric risks for some congenital disorders.
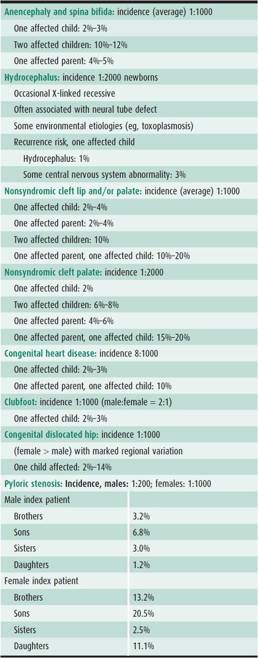
2. The recurrence risk varies with the number of affected family members. For example, after one child is born with a neural tube defect, the recurrence risk is 2%–3%. If a second affected child is born, the risk for any future child increases to 10%–12%. This is in contrast to single-gene disorders, in which the risk is the same no matter how many family members are affected.
3. The risk is higher if the defect is more severe. In Hirschsprung disease, another polygenic condition, the longer the aganglionic segment, the higher is the recurrence risk.
4. Sex ratios may not be equal. If a marked discrepancy exists, the recurrence risk is higher if a child of the less commonly affected sex has the disorder. This assumes that more genetic factors are required to raise the more resistant sex above the threshold. For example, pyloric stenosis is more common in males. If the first affected child is a female, the recurrence risk is higher than if the child is a male.
5. The risk for the offspring of an affected person is approximately the same as the risk for siblings, assuming that the spouse of the affected person has a negative family history. For many conditions, however, assortative mating, “like marrying like,” adds to risks in offspring.
NONMENDELIAN INHERITANCE
Epigenetic Regulation
Although development is regulated by genes, it is initiated and sustained by nongenetic processes. Epigenetic events are points of interaction between developmental programs and the physicochemical environments in differentiating cells. Genetic imprinting and DNA methylation are examples of epigenetic processes that affect development. Certain genes important in regulation of growth and differentiation are themselves regulated by chemical modification that occurs in specific patterns in gametes. For example, genes that are methylated are “turned off” and not transcribed. The pattern of which genes are methylated may be determined or affected by the sex of the parent of origin (see the next section). Expression of imprinted genes may sometimes be limited to specific organs (eg, the brain), and imprinting may be relaxed and methyl groups lost as development progresses. Disruption of imprinting is now recognized as contributing to birth defect syndromes (described in the next section). Certain techniques developed to assist infertile couples (advanced reproductive technology) may affect epigenetic processes and lead to genetic disorders in the offspring conceived via these methods.
Niemitz EL, Feinberg AP: Epigenetics and assisted reproductive technology: a call for investigation. Am J Hum Genet 2004;74:599 [PMID: 14991528].
Imprinting
Although the homologs of chromosome pairs may appear identical on routine karyotype analysis, it is now known that the parental origin of each homolog can affect which genes are actually transcribed and which are inactivated. The term imprinting refers to the process by which preferential transcription of certain genes takes place, depending on the parental origin, that is, which homolog (maternal or paternal) the gene is located on. Certain chromosomes, particularly chromosome X, and the autosomes 15, 11, and 7, have imprinted regions where some genes are only read from one homolog (ie, either the maternal or paternal allele) under normal circumstances, and the gene on the other homolog is normally inactivated. Errors in imprinting may arise because of uniparental disomy or UPD (in which a copy from one parent is missing), by a chromosomal deletion causing loss of the gene normally transcribed, or by mutations in the imprinting genes that normally code for transcription or inactivation of other genes downstream. A good example of how imprinting may affect human disease is Beckwith-Wiedemann syndrome, the gene for which is located on chromosome 11p15.
Cohen MM et al: Overgrowth Syndromes. New York, NY: Oxford University Press; 2002.
Genetic Anticipation
Geneticists coined the term “anticipation” to describe an unusual pattern of inheritance in which symptoms became manifest at earlier ages and with increasing severity as traits are passed to subsequent generations. Mapping of the genes responsible for these disorders led to the discovery that certain repeat sequences of DNA at disease loci were not stable when passed through meiosis. Repeated DNA sequences, in particular triplets (eg, CGG and CAG), tended to increase their copy number. As these runs of triplets expanded, they eventually affected the expression of genes and produced symptoms. Curiously, all the disorders undergoing triplet repeat expansion detected thus far produce neurologic symptoms. Most are progressive. In general, the size of the triplet expansion is roughly correlated with the timing and severity of symptoms. The reasons for the meiotic instability of these sequences are not yet understood. The mechanisms appear to involve interactions between DNA structure (eg, formation of hairpin loops) and replication enzymes (DNA polymerase complexes) during meiosis.
Triplet repeat instability can modify the inheritance of autosomal dominant, autosomal recessive, and X-linked traits. Autosomal dominant disorders include several spinal cerebellar atrophies, Huntington disease, and myotonic dystrophy. Unstable triplet repeat expansion contributes to at least one autosomal recessive disorder, Friedreich ataxia. The most common X-linked disorder demonstrating triplet repeat instability and expansion is Fragile X syndrome.
Mitochondrial Inheritance
Mitochondrial disorders can be caused by both nuclear and mitochondrial genes. The former would follow Mendelian inheritance, either AR, AD, or X-Linked, while the latter demonstrates mitochondrial inheritance. Mitochondrial DNA is double-stranded, circular, and smaller than nuclear DNA, and is found in the cytoplasm. It codes for enzymes involved in oxidative phosphorylation, which generates adenosine triphosphate. Since the 1990s, enormous advances in technology and improved clinical documentation have led to a better understanding of the interesting disorders caused by mutations in mitochondrial DNA (mtDNA).
Mitochondrial disorders can be associated with point mutations, deletions, or duplications in mtDNA. However, there is a threshold effect depending on the heteroplasmy (see next). Because of the difficulty in predicting mitochondrial DNA disorders and the variability of the clinical course, it is often difficult to calculate specific recurrence risks.
Mitochondrial disorders related to mtDNA have the following characteristics:
1. They show remarkable phenotypic variability.
2. They are maternally inherited, because only the egg has any cytoplasmic material, and during early embryo-genesis any sperm-born mitochondrial material will be eliminated.
3. In most mitochondrial disorders, cells are heteroplasmic (Figure 37–7). That is, all cells contain both normal and mutated or abnormal mtDNA. The proportion of normal to abnormal mtDNA in the mother’s egg seems to determine the severity of the offspring’s disease and the age at onset in most cases.
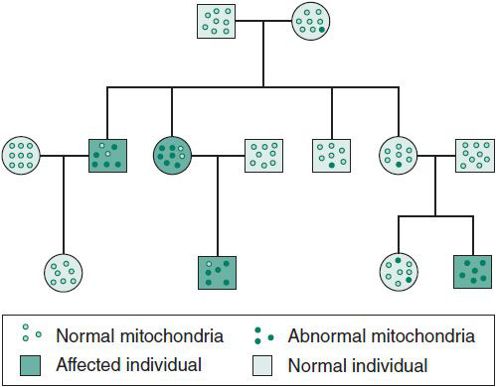
 Figure 37–7. Mitochondrial inheritance. Mutations are transmitted through the maternal line.
Figure 37–7. Mitochondrial inheritance. Mutations are transmitted through the maternal line.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


