Genetic Skeletal Disorders
John C. Carey and Michael J. Bamshad
Skeletal dysplasias are generalized disorders of bone and/or cartilage that can produce short stature, osseous deformity, and functional disabilities. More than 372 different genetic disorders of the skeleton have been described by the International Skeletal Dysplasia Society as constitutional diseases of bone1; collectively, dysplasias represent most of the intrinsic disorders of bone with the dysostoses comprising the rest. The overall frequency of the skeletal dysplasias is about 1 in 4000 births,2 making this class of disorders about as common as neurofibromatosis type 1 or Turner syndrome, conditions much better known to the pediatric practitioner.
Skeletal dysplasias have traditionally been divided into the disorders that primarily affect the growth of cartilage and bone, the chondrodystrophies (eg, achondroplasia) and disorders with primary abnormalities of bone that lead to defects in bone density or mineralization (eg, the osteogenesis imperfectas).1,3 Included in the former group are a special class of disorders caused by disorganized development of skeletal components with multiple cartilaginous exostoses, fibrodysplasia ossificans progressiva being the proteotypic syndrome. The dysostoses or disorders of bone structure that cause malformations and functional abnormalities of individual bones are also encompassed under the broader categorization of genetic skeletal disorders. Over the past decade, the distinction between these groups has become increasingly blurred and some mutations can cause abnormalities of bone and cartilage that affect various skeletal elements.1
Skeletal dysplasias can cause substantial morbidity in children and adults, yet many affected individuals lead relatively normal lives, albeit with some special challenges. Historically, the term dwarf has been used to refer to persons with bone dysplasias and disproportionate short stature. Because of the pejorative nature of this label and because it evokes thoughts of a different class of personhood, the use of the term is discouraged. The preferred terminology is to refer to a condition by its medical designation (eg, diastrophic dysplasia); however, the term dwarfism as a medical term is still widely used and accepted by many. This section describes the classification, distinguishing characteristics, and management of selected disorders.
Conventionally, the skeletal dysplasias are often grouped according to the anatomic location of the bones that are most severely affected and the histologic abnormalities that are commonly observed. For example, skeletal dysplasias that affect the spine and the epiphyses are called spondyloepiphyseal dysplasias. However, the criteria used to categorize disorders into this classification are inconsistently applied to many skeletal dysplasias (eg, achondroplasia), and this diminishes its heuristic value. The classification schemes used to organize dysostoses are quite varied. No single system has become widely adopted,1,3 and, accordingly, the clinical presentation and varied expressions of the dysostoses tend to be difficult to remember accurately.
The strategy of classifying malformations according to the developmental pathway that is disrupted can be applied to the skeletal dysplasias.1,3 The logic of the classification is to separate development of the skeleton into three primary phases: patterning, morphogenesis (ie, condensation, differentiation, and histogenesis), and growth. Dysostoses are typically produced by disturbances of skeletal patterning (malformations) in a myriad of ways that have historically been categorized by the specific bone (eg, radial aplasia, fibular-femur complex) or anatomic location affected (eg, truncation defects, posterior polydactyly). Disturbances of bone growth lead to generalized disorders of bone as exemplified by most skeletal dysplasias (eg, achondroplasia). Skeletal disorders characterized by defects of the formation of multiple individual bones have been more difficult to categorize. In these disorders, the patterning of the skeletal elements is normal, and the growth of most skeletal elements is unaffected, but the morphogenesis of particular bones is disturbed. Thus, these conditions exhibit features of both skeletal dysplasias and dysostoses. For example, most of the bones in children with campomelic dysplasia exhibit normal patterning and growth. However, the long bones (femur, tibia) of children with campomelic dysplasia are bowed and the tibiae are short because histogenesis of these bones has been perturbed. Campomelic dysplasia is caused by mutations in SOX9. The expression of the gene encoding type II collagen (COL2A1) is directly regulated by SOX9 protein and abnormal regulation of COL2A1 during chondrogenesis causes the skeletal abnormalities associated with campomelic dysplasia.
GENERAL APPROACH
The child with a skeletal dysplasia will present in primarily three ways: (1) as a newborn with short limbs or trunk in respiratory distress requiring ventilation, (2) as a newborn or older infant with disproportionate short stature, or (3) as a child with one of the various osseous manifestations associated with the bone dysplasias.4 An infant in the first group typically has one of the lethal chondrodystrophies (eg, thanatophoric dysplasia) and needs ventilatory support because of pulmonary hypoplasia or another respiratory feature of these conditions. A child with either the second or third presentation likely has one of the various disorders listed in Table 179.1. The systematic approach to the patient with any of the three presentations has been developed by Hall, Superti-Furga, and other authorities in the field (Fig. 179-1).1,3
The first step is the gathering of the history and physical examination with recognition of the decreased length or height. The pregnancy history may reveal that an abnormality was detected by ultrasonography (eg, short limbs or polyhydramnios), because prenatal diagnosis of a fetus with a presumed skeletal dysplasia is becoming more commonplace. As always, a detailed family history is important and may show relatives with short stature or consanguinity.
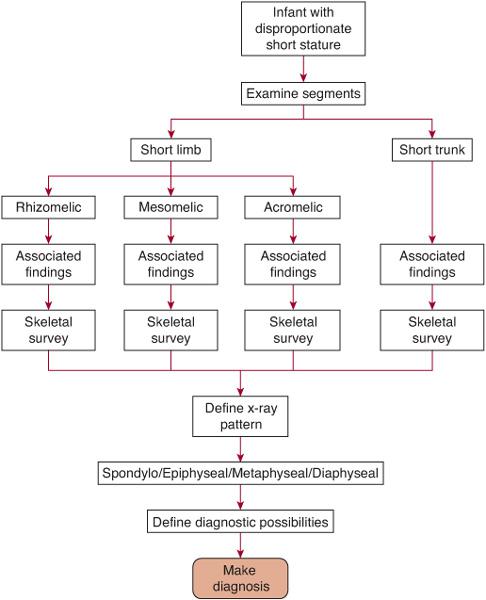
FIGURE 179-1. The diagnostic approach to the child with disproportionate short stature.
The physical examination should include accurate measurements of length/height, weight, occipito-frontal circumference (OFC), the arm span, and the upper/lower body ratio.4 The latter two measurements will define disproportion of the limbs to the trunk. The arm span is usually within 4 cm of the length/height at any age. If arm span is less than 5 cm of length, short limbs are suggested; if more than 5 cm of length, a short trunk is likely. The upper to lower ratio is taken by measuring the distance from the pubis to the heel (lower segment) and subtracting this from the length/height to obtain the upper segment. The ratio of the upper to the lower (U/L) is 1.6 to 0.93 from the newborn to the adolescent, respectively. Children with an elevated U/L ratio (eg, greater than 1.8 in a newborn) have a short-limbed form of disproportionate short stature, whereas those with a lowered ratio have a short-trunk form. Often the disproportion can be visualized just by simply looking at the infant or child; the upper limbs usually come to about one third of the length of the thigh when held down by the side. When the fingertips of the hand are at or above the iliac crest, clinical disproportion is present.
The second step in the diagnostic process involves determining which segment of a limb or trunk is the shortest. Usually in any short-limb (or short-trunk) form of short stature there is a decrease in total length. However, one portion is often more shortened than the others, and this can be a clue to a diagnosis. If the upper portion of the limb (ie, the humerus or the femur) is the shorter part (as is the case in achondroplasia), then this is referred to as rhizomelic shortening. If the middle segment of the limb (ie, forearm and lower leg) is the relatively shorter part, then this is called mesomelic shortening (as in Leri-Weil syndrome). Shortening of the distal part of the limb (ie, hands and feet) is called acromelic shortening (as in the brachydactylies). If the trunk is the predominant area of shortening (as in Morquio syndrome), then the neck, thorax, or the entire spine will be short.
The third step involves documenting all the nonskeletal physical features.4 Associated clinical findings, either malformations (eg, cleft palate, polydactyly) or important secondary findings (eg, dimples, bowing, contractures), can be helpful in leading to the diagnosis and will facilitate considering specific diagnostic paths. An important associated clinical finding is the presence or absence of serious respiratory difficulties at birth, the hallmark of the so-called lethal chondrodystrophies.
The fourth step in the process is a systematic categorization of the radiographic findings by area of involvement.4-6 A complete skeletal survey should include radiographs of the skull, long bones, antero-posterior (AP) of the pelvis, and AP/lateral of the spine and will be necessary in the evaluation of the child with a potential skeletal dysplasia. All the disorders shown in Table 179.1 are characterized by a specific pattern of skeletal abnormalities that are apparent on these radiographs. Most of the bone dysplasias have predictable and nonrandom adverse effects on the epiphyses, metaphyses, the diaphyses, and spine that vary with each condition. The architecture of the pelvic contour or the vertebral bodies is often distinctive enough to lead to a specific diagnosis (eg, thanatophoric dysplasia). Thus in this way, each skeletal dysplasia can be categorized as predominantly involving the epiphyses, metaphyses, or diaphyses and/or the spine (spondylo-). For example, if one utilizes this approach radiologically, the bone findings could be classified as showing a spondyloepiphy-seal dysplasia (Kniest dysplasia) or a spondylometaphyseal dysplasia (achondroplasia) and so on.
The final step in the evaluation, if necessary, is to confirm the clinical diagnosis using laboratory tests or histologic findings. For example, assay of skin fibroblasts for alterations of protein migration of type I collagen or direct mutation analysis of collagen from blood sampling is sometimes necessary in the diagnosis of forms of osteogenesis imperfecta. Some disorders show abnormalities of calcium and phosphorous (eg, hypophosphatemic rickets) or alkaline phosphatase (eg, hypophosphatasia) (see Chapter 542). In a lethal chondrodystrophy (eg, thanatophoric dysplasia type 1) biopsy of the growth plate at postmortem examination may be helpful in confirming a clinical diagnosis. Molecular testing for many conditions, including achondroplasia and thanatophoric dysplasia, is valuable clinically and laboratories offering such testing can be found at http://www.genetests.org.7
Once a specific diagnosis is made, a plan for health supervision and management can be organized based on the natural history of the condition. All the skeletal dysplasias, with rare exception (eg, warfarin embryopathy), are single gene disorders, and with the exception of the few X-linked conditions, are inherited in an autosomal dominant and/or recessive pattern. The risk of germ-line mosaicism is important for some conditions that are apparently new mutations (eg, osteogenesis imperfecta and campomelic dysplasia). The prenatal diagnosis of chondrodystrophies is also complicated because of changes in the technology and availability of genetic testing.
The psychological aspects of coping with the impact of a bone dysplasia are particularly unique. There are different implications for families in which average-sized parents have a child with a dysplasia or in which parents with achondroplasia (ie, heterozygotes) have a baby homo-zygous for the mutation causing achondroplasia, which is lethal. There are also different challenges for children with different conditions. In conditions such as achondroplasia or spondyloepiphy-seal dysplasia congenita, where short stature is a prominent and consistent feature, a child has to cope with the stigma of short stature, a physical appearance of disproportion, the consequences of orthopedic and neurosurgical complications, and day-to-day challenges that medical professionals rarely encounter, such as clothing and bathroom needs and practical changes around the house. The Little People of America is an outstanding resource for families and children (http://www.lpaonline.org). Most individuals with these conditions deal with them effectively and adapt their lives to these challenges. Sensitivity to the many issues surrounding the emotional and psychological impact is obviously important, and a genuine acceptance of the differences in persons with skeletal dysplasias is also crucial to developing a relationship of caring. The primary care practitioner has the opportunity to provide support in this setting and provide the medical home for the child and family.
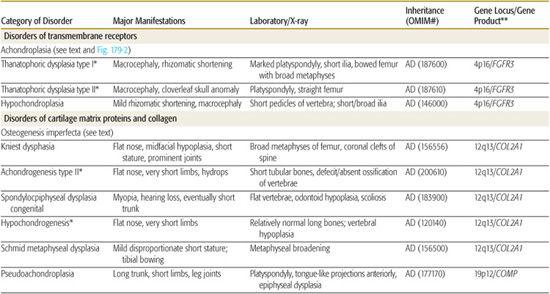
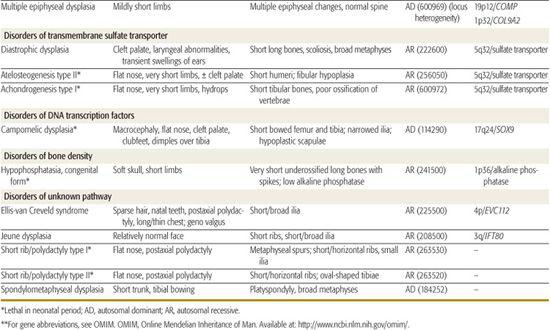
Table 179.1 lists selected skeletal dysplasias, their clinical and radiographic features, and the molecular defect (when known); the conditions are grouped by gene product and function (if known). In addition, achondroplasia and the osteogenesis imperfectas are discussed in detail. More than 50 different skeletal dysplasias that include significant respiratory insufficiency that usually results in neonatal death have been described.1,5 A neonate with disproportionate short stature who has respiratory distress may have a lethal skeletal dysplasia.
SKELETAL DYSPLASIAS CAUSED BY MUTATIONS IN TRANSMEMBRANE RECEPTORS
The prototypic conditions that involve transmembrane receptors are the achondroplasia group and result from mutations in a gene encoding a receptor (fibroblast growth factor receptor 3, FGFR3) that negatively regulates the growth of cartilage. Thus, mutations in FGFR3 activate this receptor, and as a consequence growth is significantly inhibited. The phenotypic overlap between some of the conditions in this group had been observed for decades, and thus investigators in the field were not surprised when different mutations of the same gene were discovered to cause achondroplasia, two types of thanatophoric dysplasia, and hypochondroplasia (Table 179.1).1
 ACHONDROPLASIA
ACHONDROPLASIA
Achondroplasia is the best-known skeletal dysplasia in humans, occurs in about 1 in 20,000 newborns, and is usually recognized at birth.2 The syndrome pattern consists of disproportionate short stature with rhizomelic shortening, macrocephaly, and characteristic craniofacial findings including a depressed root and nasal bridge, a prominent forehead, and midfacial hypoplasia (Fig. 179-2A). The hands are short, and the fingers are broad with digits 3 and 4 splayed more distally than proximally, giving the hand a “trident” appearance. The overall length is often in the low-average range at birth but by age 2 to 3 months, the length is below the fifth percentile. A lumbar gibbus occurs in infancy but usually resolves. Children with achondroplasia usually do not have the malformations, such as cleft palate or polydactyly, that are observed in other newborn skeletal dysplasias.
The diagnosis of achondroplasia is confirmed by the abnormalities found on the AP pelvis film that includes the upper femurs, which are quite characteristic (Fig 179-2B). The iliac bones are short and round, and the acetabulum is flattened.5,6 The shape of the ilia is similar to that found in other conditions, but the head of the femur exhibits a particularly distinctive contour. The other long bones have mildly flared metaphyses; the lumbar vertebrae have short pedicles and posterior scalloping. In general, the findings are consistent with a spondylometaphyseal dysplasia.
Individuals with achondroplasia are at risk for a number of problems and complications, including a predisposition to serous otitis media, delay in motor milestones in infancy, bowing of the legs (usually the tibia) presenting after ambulation has started, and orthodontic problems related to the maxillary hypoplasia.8,9 Growth curves are available for follow-up of the child with achondroplasia. Length and OFC can be monitored on well-child visits. Average adult height in males with achondroplasia ranges between 118 and 145 cm, and the range in females is between 112 and 136 cm. Limb-lengthening procedures have been performed on some adolescents with achondroplasia and resulted in an increase of several centimeters in height. However, this approach is controversial, and studies of long-term outcome are needed to help determine the risk/benefit ratio of this procedure.
The most important manifestation of achondroplasia is related to the stenosis of the foramen magnum and spine.8,9 The former presents in infancy, whereas the latter occurs in later years, usually adulthood. Compression of the upper cord at the foramen magnum, cervicomedullary compression, presents with a myriad of symptoms including apnea (both obstructive and central), quadriparesis, growth delays, and hydrocephalus. Any signs of compression or of hydrocephalus warrant referral to a neurosurgeon and/or neurologist. Some experts have suggested screening for compression using routine imaging, but the American Academy of Pediatrics guidelines suggest measuring the size and shape of the fontanel and monthly monitoring of the OFC. A high index of suspicion for this complication is required. Standards of the size of the foramen magnum, as measured by computed tomography (CT) or magnetic resonance imaging (MRI), in children with achondroplasia are available and can be used to help decide if there is compression at the cervicomedullary junction.8
Achondroplasia is an autosomal dominant disorder with most children having a de novo mutation of FGFR3. Most patients with achondroplasia have an identical missense mutation that results in a substitution of codon 380 of FGFR3; this missense mutation causes a glycine residue to be replaced by an arginine. Patients with hypochondroplasia and the thanatophoric dysplasias have different missense mutations in FGFR3. If needed to confirm a clinical diagnosis, molecular testing is available.7
The Little People of America (www.lpaonline.org) is a valuable resource for families of individuals with achondroplasia and all other skeletal dysplasias.
 THANATOPHORIC DYSPLASIAS
THANATOPHORIC DYSPLASIAS
Two relatively distinct skeletal dysplasias also involving mutations of FGFR3 are thanatophoric dysplasia 1 and 2, which have similar clinical characteristics but different molecular defects.1,2 Both are lethal chondrodystrophies with only a few recorded survivors beyond the neonatal period. Death is usually caused by either compression at the cervicomedullary region by the foramen magnum or pulmonary hypoplasia. The presentation is always in the newborn, with many cases now being diagnosed prenatally by ultrasound because of polyhydramnios or the detection of the short limbs.
As in achondroplasia there is true macro-cephaly; the limbs are very short with obvious disproportion. There is a notable increase in the folds of skin of the limbs and striking shortness and broadness of the digits. The radiographic findings are diagnostic with marked platyspondyly, flared metaphyses of long bones, and short iliac bones. In type 1 thanatophoric dysplasia the femurs are bowed, but in type 2 they are straight.5,6 Furthermore, the cranium of infants with type II thanatophoric dysplasia often shows the cloverleaf skull malformation.
Table 179-1. Skeletal Dysplasias
Type 1 is caused by mutations in two regions of the extracellular domain of the FGFR3 protein, whereas type 2 patients have mutations of codon 650, which is in the intracellular portion of the receptor protein. Both conditions are caused by de novo mutations of FGFR3, and thus parents are at very low recurrence risk.1 DNA testing for FGFR3 mutations is available both for prenatal diagnosis or confirmatory testing of an infant.7
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


