Genetic Eye Diseases
J. Bronwyn Bateman, John Roarty, Christina L. Butera, and Alex V. Levin
The development of techniques for identifying the genetic bases for disease has resulted in an increase in the understanding of eye diseases. The prevalence of genetic disease as a basis for significant visual loss is uncertain and dependent on the population, health care resources, and cultural values. Genetic factors may influence normal, stable development of the eye but may cause a progressive deterioration over time. Although a specific mutation of a gene may result in a consistent phenotype, variation is common and may reflect the effect of other genes or environmental factors. Complex diseases, such as myopia and strabismus, probably have both genetic and environmental bases. Population-based studies may be influenced by ascertainment bias, inconsistent data collection, or cultural factors. Autosomal recessive disorders are more common in consanguineous cultures such as occur in the Middle East or Asia. Other disorders, such as the group of diseases called retinitis pigmentosa, have a more uniform geographic prevalence rate of 20 to 40 per 100,000 worldwide. With the completion of the Human Genome Project (http://www.ornl.gov/sci/techresources/Human_Genome/home.shtml) and the rapid development of improved analytical tools, a new understanding of the genetic bases of eye diseases offers the hope of understanding causes and identifying cures.
Some ocular diseases are caused by mutations of genes that are expressed primarily or exclusively in the eye. Other disorders are multisystemic.1 A genetic basis should be identified for visual loss or malformations that are not attributable to infections or trauma to optimize both treatment of the patient and counseling of the family. The evaluation should be initiated with a careful family history and pedigree. The parents should be queried about consanguinity. It is helpful to identify the anomalies commonly encountered in genetic eye diseases and syndromes (eg, kidney abnormalities and general dysmorphic features such as abnormal interpupillary distance, epicanthal folds, and palpebral fissure slant). The ophthalmologist increasingly depends on the primary care physician to make an accurate diagnosis, and the primary care physician/geneticist depends upon the ophthalmologist for information that may be helpful in rendering a diagnosis and prognosis. Consultation with a geneticist for many conditions is wise.
This chapter will deal with some genetic diseases of the eye. Cataract, glaucoma, and corneal diseases often have a genetic basis and are discussed in Chapter 590, as are those multisystem genetic disorders that have ocular manifestations (eg, neurofibromatosis).
EMBRYOLOGY
Major structural anomalies occur in 2% to 3% of all newborns, and minor anomalies occur in 15%.2 The eye begins as a groove in the forebrain at 22 days gestation. Histologically, the lens placode is seen by 4 weeks with a lens vesicle in the optic cups by 5 weeks. The groove on the ventral surface of the optic cup closes by 7 weeks, closing the optic nerve and completing the pupil. A major regulatory gene for eye development is PAX6 (paired box gene 6), a transcription factor; other important regulatory genes are the PAX2 (paired box gene 2) and SHH (sonic hedgehog) genes. Multiple gene products function in pathways downstream of each of these genes.2 Mutations in these and other genes can lead to eye disease.
CHROMOSOMAL DISORDERS
Most chromosomal disorders are associated with abnormal eye findings, and virtually all involve other organ systems. Infants with chromosomal disorders often have multiple congenital anomalies, growth retardation, and developmental delay in addition to the ocular abnormalities. All microscopically identifiable chromosomal deletions, duplications, or aneuploidies (disorders of chromosomal numbers) are associated with some element of mental retardation, with the exception of some X-chromosomal anomalies. The ocular manifestations of chromosomal aberrations are numerous, varied, and beyond the scope of this chapter.3 Some examples of eye findings in chromosomal aberrations are listed in Table 592-1.
MONOGENIC DISORDERS
Monogenic disorders are caused by a mutation in a single gene. Some syndromes can be caused by mutations in more than one gene (genetic heterogeneity), each resulting in a similar syndromic phenotype, and may have variable inheritance patterns. Increasingly, known genes can be sequenced if the clinical diagnosis suggests a particular disease, thus allowing for a molecular diagnosis. Table 592-2 lists eye findings for many syndromic disorders. Table 592-3 provides a list of eye findings for metabolic disorders that share a similar genetic pathogenesis as the syndromes. For further discussion of inborn errors of metabolism such as the mucopolysaccharidoses, sphingolipidoses, and mucolipidoses, refer to Section 11.
GENETIC EYE DISORDERS
 MICROPHTHALMIA/ANOPHTHALMIA
MICROPHTHALMIA/ANOPHTHALMIA
Microphthalmia is a small eye (as differentiated from isolated microcornea). Anophthalmia refers to the complete absence of the eye; true anophthalmia is rare and can be confirmed only after the contents of the orbit have been examined histologically. Anophthalmia results when the optic vesicle fails to form. Primary anophthalmia may occur in an otherwise normal child or in association with a multisystem disorder such as trisomy 13, deletion of the short arm of chromosome 18, or other cerebral maldevelopment disorders. Isolated anophthalmia usually occurs sporadically and may be due to autosomal dominant, autosomal recessive, and X-linked recessive mutations.
 ANIRIDIA
ANIRIDIA
The term aniridia is a misnomer, because a small stump of iris tissue is almost always present; the disorder is rarely limited to the iris malformation (Fig. 592-1). As with most autosomal dominant disorders, expression can be extremely variable even within a family. Nystagmus (wandering or shaking eye movements), cataracts, ectopia lentis (dislocated lens), glaucoma, macular hypoplasia, optic nerve hypoplasia, and retinal dystrophy also may occur. The corneal epithelial stem cells may be abnormal, resulting in progressive vascularization of the cornea (pannus), which is associated with an increased risk of transplant rejection. Studies with stem cell transplantation and autologous serum eyedrops are ongoing. Lubrication with artificial tear preparations also may be helpful.
The “sporadic” form accounts for one third of all cases and may be associated with a deletion of the short arm of 11p13. This region includes the PAX6 and Wilms tumor genes (WT1), and absence of the intervening genes may result in genitourinary anomalies and mental retardation (WAGR syndrome—Wilms tumor; aniridia; genitourinary anomalies, usually tumors of the gonads; and mental retardation). Glaucoma frequently develops before adolescence in approximately 50% of patients. Visually significant cataracts are common. About 30% of patients with the chromosomal deletion form of aniridia develop a Wilms tumor before 3 years of age; this cancer is diagnosed before age 5 years in 80% of cases. Conversely, less than 2% of patients with Wilms tumor have aniridia. All patients with sporadic aniridia should have a karyotype, including assessment for a microdeletion using FISH or other molecular genetics techniques. Renal ultrasound study should be performed every 3 to 6 months to assess the possibility of Wilms tumor in all infants/children with sporadic aniridia, and every 3 months in those with deletions of chromosome 11.
Table 592-1. Chromosomal Aberrations and Their Eye Deformities
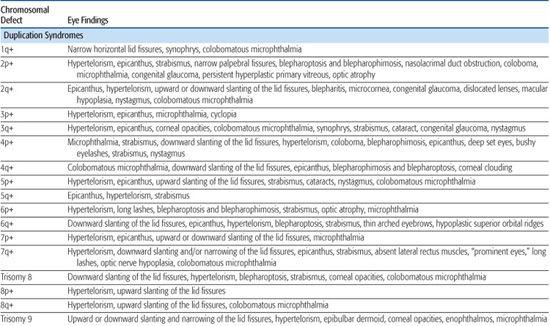

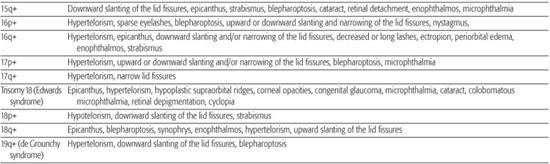

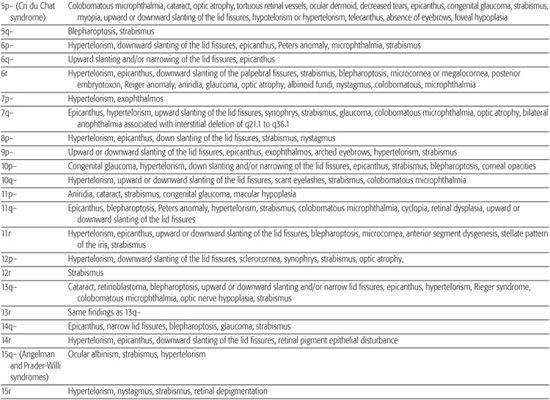
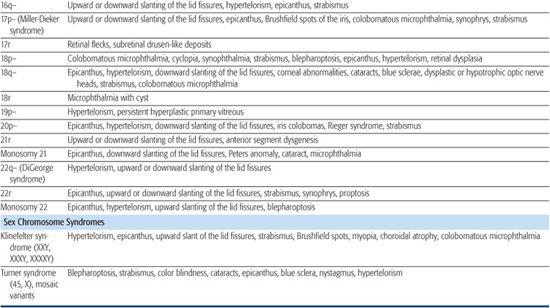
Two thirds of aniridia cases are familial, caused by mutations in the PAX6 gene and exhibit an autosomal dominant mode of transmission. Aniridia has not been reported in families with autosomal dominant Wilms tumor.
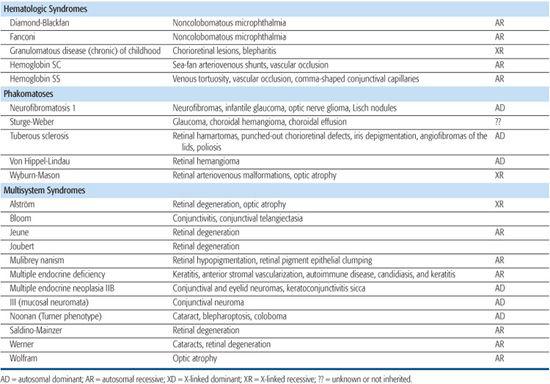
Table 592-2. Ocular Findings in Multisystem Syndromes
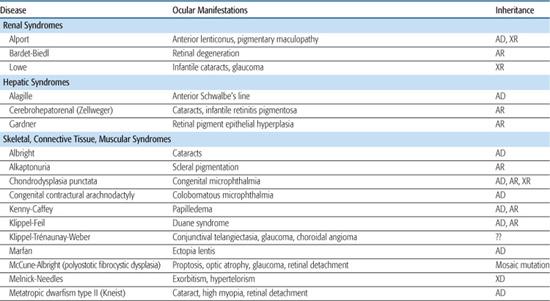
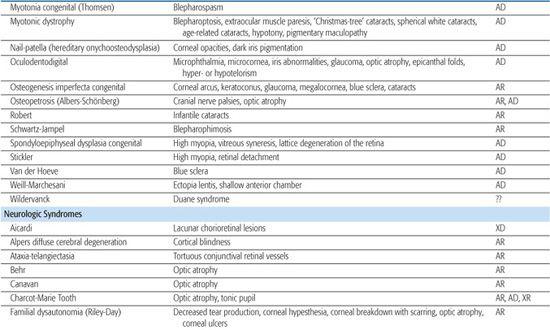
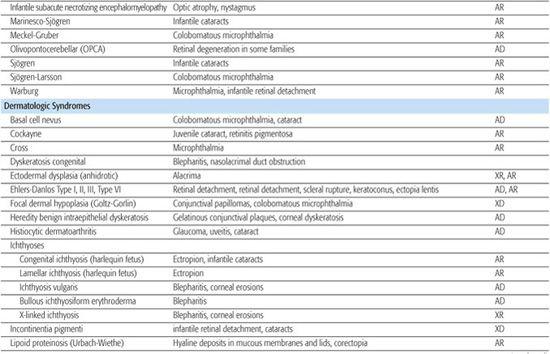
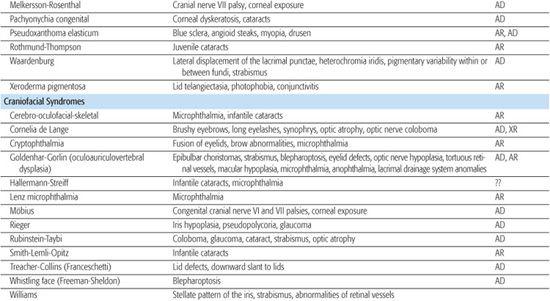
A rare autosomal recessive form of aniridia (Gillespie syndrome) has a different iris appearance and is associated with other systemic abnormalities not seen in typical dominant aniridia.
Table 592-3. Systemic Disease Associations with Retinitis Pigmentosa
Autosomal Dominant |
Alagille syndrome (arteriohepatic dysplasia) |
Charcot-Marie-Tooth disease |
Flynn-Aird syndrome |
Oculodentodigital dysplasia syndrome |
Olivopontocerebellar atrophy |
Paget disease |
Pierre Robin syndrome |
Steinert disease (myotonic dystrophy) |
Stickler syndrome |
Waardenburg syndrome |
Wagner disease |
Autosomal Recessive Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|