Ewing Sarcoma and Primitive Neuroectodermal Tumor
Crystal L. Mackall and Jeffrey A. Toretsky
Ewing sarcoma (ES) is the second most common bone tumor in children and adolescents. It was named for James Ewing, a pathologist who first described the tumor in 1921, emphasizing its distinction from osteosarcoma based upon enhanced radiosensitivity and a propensity to involve flat bones of the axial skeleton.1 In the early 1980s, both Ewing sarcoma and peripheral primitive neuroectodermal tumor (PNET) were found to contain identical t(11;22)(q24;q12) translocations.2,3 Based upon the shared translocation, and similar clinical behaviors, Ewing Sarcoma Family of Tumors (ESFT) is now considered one disease entity that includes classical ES, atypical ES, peripheral primitive neuroectodermal tumor, neuroepithelioma, and Askin tumor (an ESFT of the chest wall). It should be emphasized that peripheral PNETs are entirely different from PNETs arising in the central nervous system which do not bear the t(11;22) translocation and are not considered part of the ESFT category.
 EPIDEMIOLOGY, PATHOPHYSIOLOGY, AND GENETICS
EPIDEMIOLOGY, PATHOPHYSIOLOGY, AND GENETICS
ESFTs occur in approximately 2 to 3 per million/year in persons less than 20 years4 with a peak in the second decade of life, although they have been reported in infants and occur with a substantial incidence in adults, especially prior to age 40. For unknown reasons, ESFT are extremely rare among individuals of African and Asian heritage. 
The cell of origin that gives rise to ESFT is also unknown, with primitive neural cells implicated historically, and recent studies suggesting that mesenchymal stem cells may give rise to the disease.6,7 The classic translocation t(11;22)(q24;q12), or another related translocation, occurs in greater than 95% of ESFT.8 ESFT-associated translocations join the Ewing sarcoma (EWS) gene located on chromosome 22 to an ets-family gene, most commonly FLI1 (Friend Leukemia Insertion), located on chromosome 11.9 The 68-kDa protein produced by the EWS-FLI1 fusion transcript functions as an aberrant transcription factor and plays a critical role in initiating and sustaining ESFT. Expression of this molecule can transform mouse fibroblasts,10 and reduction of EWS-FLI1 expression induces death of ESFT cell lines.11-13 Such studies have established the EWS-ets translocation as a compelling therapeutic target. However, clinically applicable therapies to successfully prevent expression of EWS-ets or interfere with downstream oncogenic events induced by the fusion transcript have proven elusive thus far.14
 CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
CLINICAL FEATURES AND DIFFERENTIAL DIAGNOSIS
ESFTs most often arise in bone and have been reported in virtually every bone, but can also occur in soft tissues without bony involvement. Unlike osteosarcoma, which usually arises in the metaphysis of long bones, ESFT arises more frequently in flat bones (eg, pelvis, vertebrae, ribs, skull), and when it arises in long bones, it commonly involves the midshaft or diaphysis. ESFT patients usually present with pain or a palpable mass, and ESFT should be included in the differential diagnosis of any bone or soft tissue mass in patients from infancy through early adulthood. Unfortunately, many patients ultimately diagnosed with ESFT often describe a history of chronic pain for many months before the correct diagnosis is made. This is especially true for pelvic ESFTs when a palpable mass is not present and the presenting symptoms of neuropathic, radicular pain or vague back discomfort are often attributed to sports-related or other trivial injuries in adolescents and young adults. It is important, therefore, that children and young adults who experience persistent or recurring pelvic, back, or radicular pain undergo appropriate radiographic evaluation to rule out a malignancy involving the spine, pelvis, or extremity. Radionuclide bone scan may be particularly helpful in identifying a neoplastic cause of chronic pelvic, back, or radicular pain. Occasionally, ESFTs present with vertebral involvement, and in such cases, an associated extradural mass can compress the spinal cord and induce paraparesis or paraplegia, with sphincter dysfunction. Tumors adjacent to the spinal canal require immediate magnetic resonance imaging (MRI) scanning because intervention is sometimes needed to prevent neurologic deterioration, which can occur rapidly if spinal cord compression is present.
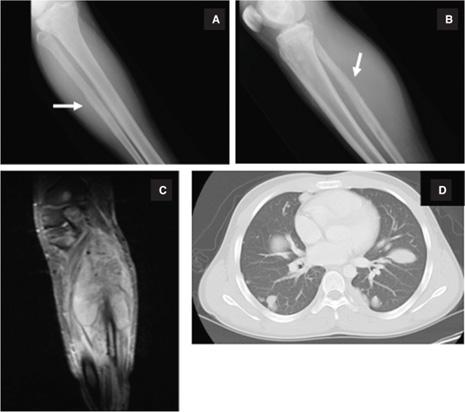
Classic radiologic findings of ESFT include permeative bone destruction with a moth-eaten appearance, elevation of periosteum with periosteal reaction (Codman triangle) or a lamellar (onion skin) lesion on plain radiographs, which results from cortical destruction followed by reparative cortical bone laid down by reactive osteoblasts (Fig. 454-1). Computerized tomography (CT) scans provide optimal imaging to assess the degree of cortical bone involvement, whereas MRI accurately establishes the extent of soft tissue and marrow involvement (Fig. 454-1). The differential diagnosis of destructive bony lesions should include osteogenic sarcoma, other sarcomas arising in bone (malignant fibrous histiocytoma, chondrosarcoma, fibrosarcoma), giant cell tumors, benign neoplasms, eosinophilic granuloma, lymphoma, or metastases from another primary neoplasm. For patients with a soft tissue mass without bony involvement, the differential diagnosis includes rhabdomyosarcoma, neuroblastoma, other soft-tissue sarcomas, lymphoma, or benign neoplasms. Systemic symptoms of fever and weight loss can occur as part of the presenting symptom complex in patients presenting with ESFT, especially with large primary tumors or metastatic disease.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


