Endocrine Neoplasia Syndromes
Steven D. Chernausek and Constantine A. Stratakis
INTRODUCTION
Most endocrine tumors in children occur in the context of genetic conditions predisposing to multiple neoplasias: multiple endocrine neoplasia types 1 and 2 (MEN-1 and MEN-2), Mc-Cune Albright syndrome, Carney complex, Von Hippel-Lindau (VHL) disease, Peutz-Jeghers syndrome (PJS), Cowden disease (CD), hereditary hyperparathyroidism and jaw tumor syndrome (HPJTS), and other extraordinarily rare conditions such as the isolated paraganglioma and Carney-Stratakis syndromes, Carney triad, Burt-Hogg-Dubé, and others. Therefore, it is now essential that the evaluation and management of these patients involves experts in cancer genetics and includes formal genetic counseling. Gene testing may be offered but performed in the setting of a cancer genetic consultation which includes pretest and posttest counseling. If a family-specific mutation is found, the genetics consultant can offer mutation-specific predictive testing to relatives. A list of endocrine neoplasia syndromes and their genetic causes is provided in Table 537-1. Disease associations in the multiple endocrine neoplasia syndromes are shown in Table 537-2. In this chapter we will discuss only the MEN conditions and the related Carney complex. Pheochromocytoma is also discussed as an endocrine tumor common in a number of endocrine neoplasia syndromes.
MULTIPLE ENDOCRINE NEOPLASIA SYNDROMES
Multiple endocrine neoplasia (MEN) syndromes as well as related conditions (such as Carney complex) are autosomal dominant disorders in which specific endocrine glands and other organs develop hyperplasia or neoplasia.1,2 Though more often seen in adults, these conditions may present in childhood. For MEN 2 and the familial pheochromocytomas, it is extremely important to diagnose these genetic conditions during childhood in order to provide periodic screening for the development of neoplasia and to offer timely prophylactic surgery.3,4
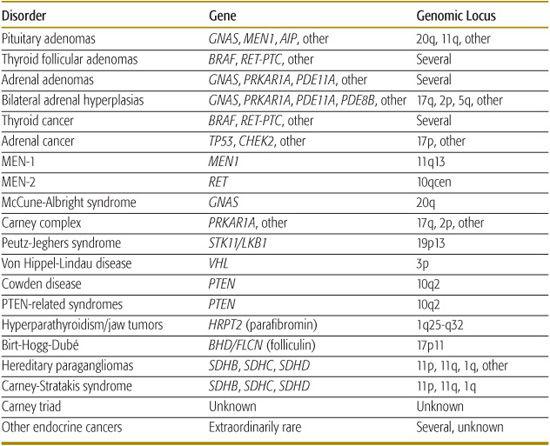
Table 537-1. Endocrine and Other Tumors in Childhood and Young Adulthood and Their Genetic Causes
 MULTIPLE ENDOCRINE NEOPLASIA 1 (MEN 1)
MULTIPLE ENDOCRINE NEOPLASIA 1 (MEN 1)
Pathophysiology
MEN 1 is caused by germline mutations in MEN1. MEN1 encodes MENIN, which functions as a tumor suppressor.2,5 The principal glands affected in MEN 1 are the parathyroid, pituitary, and pancreatic islets.6 Nearly all individuals who harbor mutations characteristic of MEN 1 develop clinically significant disease at some point in their lifetime.7
Clinical Features
Hyperparathyroidism is the most common endocrine abnormality and is characterized by disease in multiple glands.1,2 Once manifest, it typically progresses slowly and steadily over time. The next most common is intrapancreatic islet cell tumors (gastrinomas and β cell tumors). Carcinoid tumors, adrenal cortical hyperplasia, and lipomas are also found in these individuals. Collagenomas and facial angiomata have been found in up to 90% of the patients with MEN 1 (Fig. 537-1A, B). These cutaneous features can suggest carrier status within affected families.1
Genetic Screening
Because of a nearly inevitable appearance of serious endocrine abnormalities, affected families should undergo screening to detect clinical manifestations before complications occur from hormone excess or tumors metastasis. Genetic testing should be offered to individuals with familial endocrinopathies typical for MEN 1 and should also be considered in sporadic cases where 2 or more MEN-1-type tumors occur. An abnormality in the MEN1 gene can be found in approximately 70% of cases of typical MEN 1. These include a wide variety of point mutations and deletions that result in inactivation of the MEN1 gene (eFig. 537.1C  ). If a specific mutation is identified, molecular genetic methods can determine carrier status. Most of the other cases probably also have defects that affect MENIN expression but were undetected by genetic analysis.7 Genetic testing should be offered to individuals with familial endocrinopathies typical for MEN 1. It should also be considered in sporadic cases where there are 2 or more MEN-1-type tumors.
). If a specific mutation is identified, molecular genetic methods can determine carrier status. Most of the other cases probably also have defects that affect MENIN expression but were undetected by genetic analysis.7 Genetic testing should be offered to individuals with familial endocrinopathies typical for MEN 1. It should also be considered in sporadic cases where there are 2 or more MEN-1-type tumors.
Management
Current recommendations for carriers include annual biochemical monitoring for hyperparathyroidism, hyperinsulinism, and anterior pituitary disease beginning at age 5 years, accompanied by less frequent imaging studies. Testing for gastrinoma (which manifests as ulcers in the Zollinger-Ellison syndrome), carcinoid, and other intrapancreatic tumors should begin after age 20 years. This periodic screening will hopefully increase the early detection of tumors and will likely improve long-term survival, but this has yet to be proven effective.
Treatment of the hyperparathyroidism of MEN 1 involves subtotal parathyroidectomy with removal of 3.5 glands or a total parathyroidectomy with reimplantation.1 Similarly, when dealing with β-cell disease, the likelihood of multiple insulinomas is high and partial pancreatectomy might be required.
 MULTIPLE ENDOCRINE NEOPLASIA 2 (MEN 2)
MULTIPLE ENDOCRINE NEOPLASIA 2 (MEN 2)
Pathophysiology
The MEN 2 syndromes include MEN 2A, MEN 2B, and familial medullary thyroid carcinoma (FMTC) (eFig. 537.2A  ). MEN 2 is caused by gain of function mutations in the RET proto-oncogene (eFig. 537.2B
). MEN 2 is caused by gain of function mutations in the RET proto-oncogene (eFig. 537.2B  ). The RET proto-oncogene encodes RET protein, which is a cell surface growth factor receptor.8,9 It mediates the actions of the glial-derived neurotrophic factor family of peptide growth factors.10 These factors regulate neural tissue development in peripheral and enteric nervous systems.11 In contrast to the situation in MEN 1 where a wide variety of specific mutations cause disease, a much smaller number of genetic mutations cause MEN 2.1 This is because these are activating mutations as opposed to inactivating mutations. Furthermore, the specific mutations observed correlate with the phenotype of the patients and with forms and aggressiveness of the associated neoplasms.12 For example, codon 634 mutations almost always produce MEN 2A, whereas mutations more toward the N-terminus of the protein typically produce FMTC. For MEN 2B, 95% of the patients have a mutation in the intracellular tyrosine kinase domain (M918T).
). The RET proto-oncogene encodes RET protein, which is a cell surface growth factor receptor.8,9 It mediates the actions of the glial-derived neurotrophic factor family of peptide growth factors.10 These factors regulate neural tissue development in peripheral and enteric nervous systems.11 In contrast to the situation in MEN 1 where a wide variety of specific mutations cause disease, a much smaller number of genetic mutations cause MEN 2.1 This is because these are activating mutations as opposed to inactivating mutations. Furthermore, the specific mutations observed correlate with the phenotype of the patients and with forms and aggressiveness of the associated neoplasms.12 For example, codon 634 mutations almost always produce MEN 2A, whereas mutations more toward the N-terminus of the protein typically produce FMTC. For MEN 2B, 95% of the patients have a mutation in the intracellular tyrosine kinase domain (M918T).
Table 537-2. Disease Associations in the Multiple Endocrine Neoplasia Syndromes
Multiple Endocrine Neoplasia Type 1 |
Parathyroid hyperplasia or adenoma |
Islet cell hyperplasia, adenoma, or carcinoma |
Pituitary hyperplasia or adenoma |
Less common: carcinoid, pheochromocytoma, subcutaneous or visceral lipomas, cutaneous lichen amyloidosis |
Multiple Endocrine Neoplasia Type 2 |
Type 2a |
Medullary thyroid carcinoma |
Pheochromocytoma |
Parathyroid hyperplasia or adenoma |
Familial medullary carcinoma |
Type 2b |
Medullary thyroid carcinoma |
Pheochromocytoma |
Mucosal and gastrointestinal neuromas |
Intestinal neuronal dysplasia (presents like Hirschsprung disease) |
Marfanoid features |
Mixed Syndromes Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|