FEEDBACK CONTROL OF HORMONE SECRETION
Hormone secretion is regulated, for the most part, by feedback in response to changes in the internal environment. When the metabolic imbalance is corrected, stimulation of the hormone secretion ceases and may even be inhibited. Overcorrection of the imbalance stimulates secretion of a counterbalancing hormone or hormones, so that homeostasis is maintained within relatively narrow limits.
Hypothalamic-pituitary control of hormonal secretion is also regulated by feedback. End-organ failure (endocrine gland insufficiency) leads to decreased circulating concentrations of endocrine gland hormones and thence to increased secretion of the respective hypothalamic releasing and pituitary hormones (see Table 34–1; Figure 34–1). If restoration of normal circulating concentration of hormones occurs, feedback inhibition at the pituitary and hypothalamus results in cessation of the previously stimulated secretion of releasing and pituitary hormones and restoration of their circulating concentrations to normal.
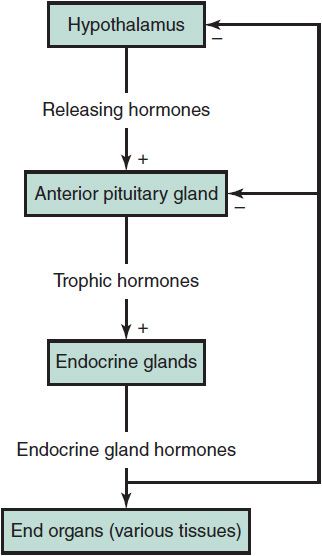
 Figure 34–1. General scheme of the hypothalamus-pituitary-endocrine gland axis. Releasing hormones synthesized in the hypothalamus are secreted into the hypophyseal portal circulation. Trophic hormones are secreted by the pituitary gland in response, and they in turn act on specific endocrine glands to stimulate the secretion of their respective hormones. The endocrine gland hormones exert their respective effects on various target tissues (end organs) and exert a negative feedback (feedback inhibition) on their own secretion by acting at the level of the pituitary and hypothalamus. This system is characteristic of those hormones listed in Table 34–1 (third level).
Figure 34–1. General scheme of the hypothalamus-pituitary-endocrine gland axis. Releasing hormones synthesized in the hypothalamus are secreted into the hypophyseal portal circulation. Trophic hormones are secreted by the pituitary gland in response, and they in turn act on specific endocrine glands to stimulate the secretion of their respective hormones. The endocrine gland hormones exert their respective effects on various target tissues (end organs) and exert a negative feedback (feedback inhibition) on their own secretion by acting at the level of the pituitary and hypothalamus. This system is characteristic of those hormones listed in Table 34–1 (third level).
Similarly, if there is autonomous endocrine gland hyperfunction (eg, McCune-Albright syndrome, Graves disease, or adrenal tumor), the specific hypothalamic releasing and pituitary hormones are suppressed (see Figure 34–1).
Bethin K, Fuqua JS: General concepts and physiology. In: Kappy MS, Allen DB, Geffner ME (eds): Pediatric Practice-Endocrinology. McGraw Hill; 2010:1–22.
DISTURBANCES OF GROWTH
Disturbances of growth and development are the most common problems evaluated by a pediatric endocrinologist. While most cases represent normal developmental variants, it is critical to identify abnormal growth patterns, as deviations from the norm can be the first or only manifestation of an endocrine disorder. Height velocity is the most critical parameter in evaluating a child’s growth. A persistent increase or decrease in height percentiles between age 2 years and the onset of puberty always warrants evaluation. Similarly, substantial deviations from target height may be indications of underlying endocrine or skeletal disorders. It is more difficult to distinguish normal from abnormal growth in the first 2 years of life, as infants may have catch-up or catch-down growth during this period. Similarly, the variable timing of the onset of puberty makes early adolescence another period during which evaluation of growth abnormalities may require careful consideration.
Appropriate standards must be used to evaluate growth. The National Center for Health Statistics provides standard cross-sectional growth charts for North American children (see Chapter 2) and the World Health Organization (WHO) growth charts use an ethnically more diverse sample. Normal growth standards may vary with country of origin. Growth charts are available for some ethnic groups in North America and for some syndromes with specific growth disturbance such as Turner or Down syndromes.
TARGET HEIGHT & SKELETAL MATURATION
A child’s growth and height potential is determined largely by genetic factors. The target (midparental) height of a child is calculated from the mean parental height plus 6.5 cm for boys or minus 6.5 cm for girls. This calculation helps identify a child’s genetic growth potential. Most children achieve an adult height within 8 cm of the midparental height. Another parameter that determines growth potential is skeletal maturation or bone age. Beyond the neonatal period, bone age is evaluated by comparing a radiograph of the child’s left hand and wrist with the standards of Greulich and Pyle. Delayed or advanced bone age is not diagnostic of any specific disease, but the extent of skeletal maturation allows determination of remaining growth potential as a percentage of total height and allows prediction of ultimate height.
SHORT STATURE
It is important to distinguish normal variants of growth (familial short stature and constitutional growth delay) from pathologic conditions (Table 34–2). Pathologic short stature is more likely in children whose growth velocity is abnormal (crossing major height percentiles on the growth curve) or who are significantly short for their family. Children with chronic illness or nutritional deficiencies may have poor linear growth, but this is typically associated with inadequate weight gain. In contrast, endocrine causes of short stature are usually associated with normal or excessive weight gain.
Table 34–2. Causes of short stature.

1. Familial Short Stature & Constitutional Growth Delay
Children with familial short stature typically have normal birth weight and length. In the first 2 years of life, their linear growth velocity decelerates as they near their genetically determined percentile. Once the target percentile is reached, the child resumes normal linear growth parallel to the growth curve, usually between 2 and 3 years of age. Skeletal maturation and timing of puberty are consistent with chronologic age. The child grows along his/her own growth percentile and the final height is short but appropriate for the family (Figure 34–2). For example, an infant boy of a mother who is 5 ft 0 in and father who is 5 ft 5 in (calculated midparental height 5 ft 5 in) may have a birth length at the 50th percentile. However, this child’s length percentile will drift downward during the first 2 years of life and will settle in at the fifth percentile where it will stay.
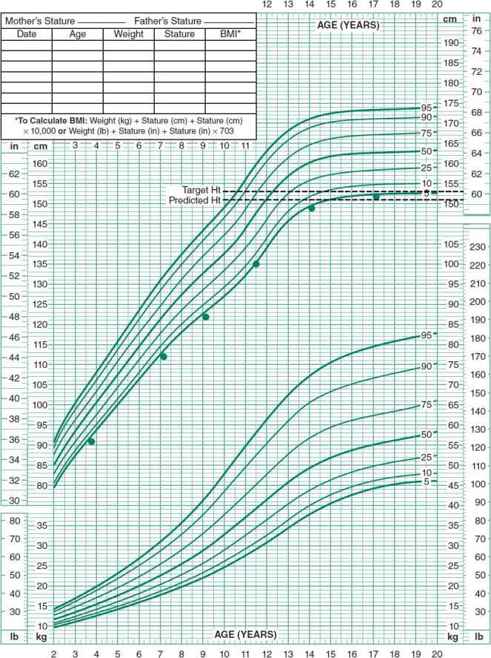
 Figure 34–2. Typical pattern of growth in a child with familial short stature. After attaining an appropriate percentile during the first 2 years of life, the child will have normal linear growth parallel to the growth curve. Skeletal maturation and the timing of puberty are consistent with chronologic age. The height percentile the child has been following is maintained, and final height is short but appropriate for the family.
Figure 34–2. Typical pattern of growth in a child with familial short stature. After attaining an appropriate percentile during the first 2 years of life, the child will have normal linear growth parallel to the growth curve. Skeletal maturation and the timing of puberty are consistent with chronologic age. The height percentile the child has been following is maintained, and final height is short but appropriate for the family.
Children with constitutional growth delay do not necessarily have short parents but have a growth pattern similar to those with familial short stature. The difference is that children with constitutional growth delay have a delay in skeletal maturation and a delay in the onset of puberty. In these children, growth continues beyond the time the average child stops growing, and final height is appropriate for target height (Figure 34–3). There is often a history of other family members being “late bloomers.”
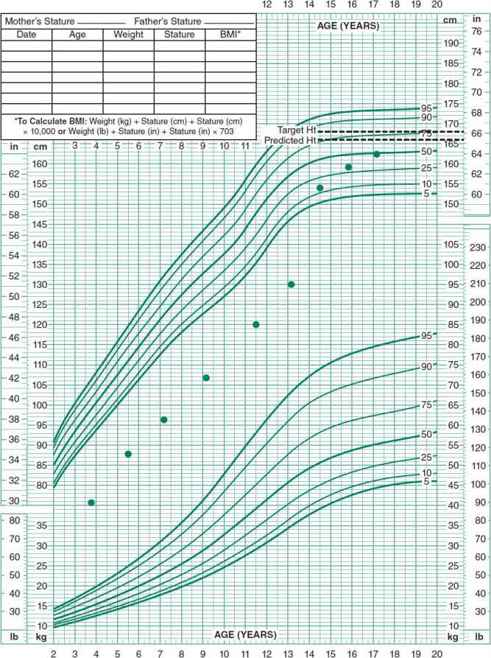
 Figure 34–3. Typical pattern of growth in a child with constitutional growth delay. Growth slows during the first 2 years of life, similarly to children with familial short stature. Subsequently the child will have normal linear growth parallel to the growth curve. However, skeletal maturation and the onset of puberty are delayed. Growth continues beyond the time the average child has stopped growing, and final height is appropriate for target height.
Figure 34–3. Typical pattern of growth in a child with constitutional growth delay. Growth slows during the first 2 years of life, similarly to children with familial short stature. Subsequently the child will have normal linear growth parallel to the growth curve. However, skeletal maturation and the onset of puberty are delayed. Growth continues beyond the time the average child has stopped growing, and final height is appropriate for target height.
2. Growth Hormone Deficiency
Human growth hormone (GH) is produced by the anterior pituitary gland. Secretion is stimulated by growth hormone–releasing hormone (GHRH) and inhibited by somatostatin. GH is secreted in a pulsatile pattern in response to sleep, exercise, and hypoglycemia and has direct growth-promoting and metabolic effects (Figure 34–4). GH also promotes growth indirectly by stimulating production of insulin-like growth factors, primarily IGF-1.
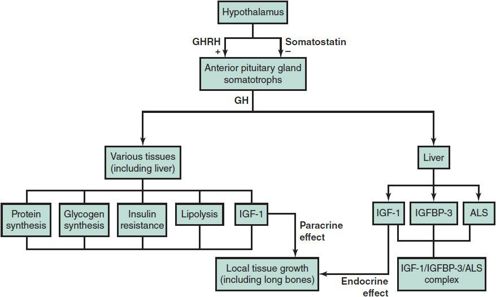
 Figure 34–4. The GHRH/GH/IGF-1 system. The effects of GH on growth are partly due to its direct anabolic effects in muscle, liver, and bone. In addition, GH stimulates many tissues to produce IGF-1 locally, which stimulates the growth of the tissue itself (paracrine effect of IGF-1). The action of GH on the liver results in the secretion of IGF-1 (circulating IGF-1), which stimulates growth in other tissues (endocrine effect of IGF-1). The action of growth hormone on the liver also enhances the secretion of IGF-binding protein 3 (IGFBP-3) and acid-labile subunit (ALS), which form a high-molecular-weight complex with IGF-1. The function of this complex is to transport IGF-1 to its target tissues, but the complex also serves as a reservoir and possible inhibitor of IGF-1 action. In various chronic illnesses, the direct metabolic effects of GH are inhibited; the secretion of IGF-1 in response to GH is blunted, and in some cases IGFBP-3 synthesis is enhanced, resulting in marked inhibition in the growth of the child. IGF-1, insulin-like growth factor 1; GH, growth hormone; GHRH, growth hormone–releasing hormone.
Figure 34–4. The GHRH/GH/IGF-1 system. The effects of GH on growth are partly due to its direct anabolic effects in muscle, liver, and bone. In addition, GH stimulates many tissues to produce IGF-1 locally, which stimulates the growth of the tissue itself (paracrine effect of IGF-1). The action of GH on the liver results in the secretion of IGF-1 (circulating IGF-1), which stimulates growth in other tissues (endocrine effect of IGF-1). The action of growth hormone on the liver also enhances the secretion of IGF-binding protein 3 (IGFBP-3) and acid-labile subunit (ALS), which form a high-molecular-weight complex with IGF-1. The function of this complex is to transport IGF-1 to its target tissues, but the complex also serves as a reservoir and possible inhibitor of IGF-1 action. In various chronic illnesses, the direct metabolic effects of GH are inhibited; the secretion of IGF-1 in response to GH is blunted, and in some cases IGFBP-3 synthesis is enhanced, resulting in marked inhibition in the growth of the child. IGF-1, insulin-like growth factor 1; GH, growth hormone; GHRH, growth hormone–releasing hormone.
Growth hormone deficiency (GHD) is characterized by decreased growth velocity and delayed skeletal maturation in the absence of other explanations. Laboratory tests indicate subnormal GH secretion or action. GHD may be isolated or coexist with other pituitary hormone deficiencies and may be congenital (septo-optic dysplasia or ectopic posterior pituitary), genetic (GH or GHRH gene mutation), or acquired (craniopharyngioma, germinoma, histiocytosis, or cranial irradiation). Idiopathic GHD is the most common deficiency state with an incidence of about 1:4000 children. Patients have also been described with a GH resistance syndrome caused by mutations in the GH receptor or other components of the GH signaling pathway. The presentation of GH resistance is similar to that of GHD, but short stature is often severe, with little or no response to GH therapy. Some mutations, such as Laron dwarfism, are accompanied by facial and skeletal dysmorphology.
Infants with GHD have normal birth weight with only slightly reduced length, suggesting that GH is a minor contributor to intrauterine growth. GH deficient infants may present with hypoglycemia, particularly when associated with other pituitary deficiencies such as central adrenal insufficiency. Micropenis may be a feature of newborn males with gonadotropin and GH deficiency. Isolated GHD and hypopituitarism may be unrecognized until late in infancy or childhood as growth retardation may be delayed until later childhood. Regardless of onset, the primary manifestation of idiopathic or acquired GHD is subnormal growth velocity (Figure 34–5). Because GH promotes lipolysis, many GH deficient children have excess truncal adiposity.
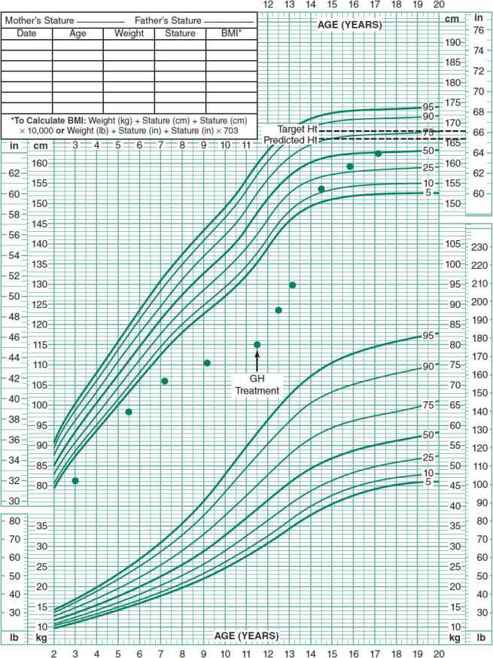
 Figure 34–5. Typical pattern of growth in a child with acquired growth hormone deficiency (GHD). Children with acquired GHD have an abnormal growth velocity and fail to maintain height percentile during childhood. Other phenotypic features (central adiposity and immaturity of facies) may be present. Children with congenital GHD will cross percentiles during the first 2 years of life, similarly to the pattern seen in familial short stature and constitutional delay, but will fail to attain a steady height percentile subsequently.
Figure 34–5. Typical pattern of growth in a child with acquired growth hormone deficiency (GHD). Children with acquired GHD have an abnormal growth velocity and fail to maintain height percentile during childhood. Other phenotypic features (central adiposity and immaturity of facies) may be present. Children with congenital GHD will cross percentiles during the first 2 years of life, similarly to the pattern seen in familial short stature and constitutional delay, but will fail to attain a steady height percentile subsequently.
Laboratory tests to assess GH status may be difficult to interpret. Children with normal short stature have a broad range of GH secretion patterns and there is significant overlap between normal and GH deficient children. Random samples for measurement of serum GH are of no value in the diagnosis of GHD, as GH secretion is pulsatile. Serum concentrations of IGF-1 give reasonable estimations of GH secretion and action in the adequately nourished child (see Figure 34–4), and are often used as a first step in the evaluation for GHD. IGF-binding protein 3 (IGFBP-3) is a much less sensitive marker of GH deficiency, but may be useful in the underweight child or in children under 4 years of age, since it is less affected by age or nutritional status. Provocative studies using such agents as insulin, arginine, levodopa, clonidine, or glucagon are traditionally done to clarify GH secretion, but are not physiologic and are often poorly reproducible, ultimately limiting their value in the clarification of GH secretion. When results of GH tests are equivocal and the clinical suspicion very high, a trial of GH treatment may help determine whether an abnormally short child will benefit from GH. Currently, the recommended treatment schedule for GHD is subcutaneous recombinant GH given subcutaneously 7 days per week with total weekly dose of 0.15–0.3 mg/kg.
GH therapy is approved by the U.S. Food and Drug Administration (FDA) for children with GHD and growth restriction associated with chronic renal failure, for girls with Turner syndrome, children with Prader-Willi and Noonan’s syndromes, and children born small for gestational age (SGA) who fail to demonstrate catch-up growth by age 4. Dose ranges for these non-GHD indications are generally different than for GHD (lower doses with Prader-Willi syndrome, higher doses with Turner syndrome and SGA). GH therapy has also been approved for children with idiopathic short stature whose current height is more than 2.25 standard deviations below the normal range for age. Final height may be 5–7 cm taller in this population. This last indication is controversial and the role of GH for idiopathic short stature is still unclear, especially due to the expense, long duration of treatment, and unclear psychological consequences. Side effects of recombinant GH are uncommon but include benign intracranial hypertension and slipped capital femoral epiphysis. With early diagnosis and treatment, children with GHD reach normal or near-normal adult height. Recombinant IGF-1 injections may be used to treat children with GH resistance or IGF-1 deficiency, but improvements in growth are not as substantial as seen with GH therapy for GH deficiency.
3. Small for Gestational Age/Intrauterine Growth Restriction
SGA infants have birth weights below the 10th percentile for the population’s birth weight–gestational age relationship. SGA infants include constitutionally small infants and infants with intrauterine growth restriction (IUGR).
SGA/IUGR may be a result of poor maternal environment, intrinsic fetal abnormalities, congenital infections, or fetal malnutrition. Intrinsic fetal abnormalities causing SGA/IUGR (often termed primordial short stature) include Russell-Silver, Seckel, Noonan, Bloom, and Cockayne syndromes, and progeria. Many children with mild SGA/IUGR and no intrinsic fetal abnormalities exhibit catch-up growth during the first 3 years. However, 15%–20% remain short throughout life, particularly those whose growth restriction in utero occurred over more than just the last 2–3 months of gestation. Catch-up growth may also be inadequate in preterm SGA/IUGR infants with poor postnatal nutrition. Children who do not show catch-up growth may have normal growth velocity, but follow a lower height percentile than expected for the family. In contrast to children with constitutional growth delay, those with SGA/IUGR have skeletal maturation that corresponds to chronologic age or is only mildly delayed.
GH therapy for SGA/IUGR children with growth delay is FDA approved and appears to increase growth velocity and final adult height.
4. Disproportionate Short Stature
There are more than 200 sporadic and genetic skeletal dysplasias that may cause disproportionate short stature. Measurements of arm span and upper-to-lower body segment ratio are helpful in determining whether a child has normal body proportions. If disproportionate short stature is found, a skeletal survey may be useful to detect specific radiographic features characteristic of some disorders. The effect of GH on most of these rare disorders is unknown.
5. Short Stature Associated With Syndromes
Short stature is associated with many syndromes, including Turner, Down, Noonan, and Prader-Willi. Girls with Turner syndrome often have recognizable features such as micrognathia, webbed neck, low posterior hairline, edema of hands and feet, multiple pigmented nevi, and an increased carrying angle. However, short stature can be the only obvious manifestation of Turner syndrome. Consequently, any girl with unexplained short stature for family warrants a chromosomal evaluation. Although girls with Turner syndrome are not usually GH deficient, GH therapy can improve final height by an average of 6.0 cm. Duration of GH therapy is a significant predictor of long-term height gain; consequently, it is important that Turner syndrome be diagnosed early and GH started as soon as possible.
GH is approved for treatment of growth failure in Prader-Willi syndrome. Many affected individuals are GH deficient and GH improves growth, body composition, and physical activity. A few deaths have been reported in Prader-Willi children receiving GH, all of which occurred in very obese children, children with respiratory impairments, sleep apnea, or unidentified respiratory infections. The role of GH, if any, in these deaths is unknown. However, as a precaution, it is recommended that all Prader-Willi patients be evaluated for upper airway obstruction and sleep apnea prior to starting GH therapy.
Children with Down syndrome should be evaluated for GHD only if their linear growth is abnormal compared with the Down syndrome growth chart.
6. Psychosocial Short Stature (Psychosocial Dwarfism)
Psychosocial short stature refers to growth retardation associated with emotional deprivation. Undernutrition probably contributes to growth retardation in some of these children. Other symptoms include unusual eating and drinking habits, bowel and bladder incontinence, social withdrawal, and delayed speech. GH secretion in children with psychosocial short stature is diminished, but GH therapy is usually not beneficial. A change in the psychological environment at home usually results in improved growth and improvement of GH secretion, personality, and eating behaviors.
 Clinical Evaluation
Clinical Evaluation
Laboratory investigation should be guided by the history and physical examination. Data included in history and physical include history of chronic illness and medications, birth weight and height, pattern of growth since birth, familial growth patterns, pubertal stage, dysmorphic features, body segment proportion, and psychological health. In a child with poor weight gain as the primary disturbance, a nutritional assessment is indicated. The following laboratory tests may be useful as guided by history and clinical judgment:
1. Radiograph of left hand and wrist for bone age
2. Complete blood count (to detect chronic anemia or leukocyte markers of infection)
3. Erythrocyte sedimentation rate (often elevated in collagen-vascular disease, cancer, chronic infection, and inflammatory bowel disease)
4. Urinalysis, blood urea nitrogen, and serum creatinine (occult renal disease)
5. Serum electrolytes, calcium, and phosphorus (renal tubular disease and metabolic bone disease)
6. Stool examination for fat, or measurement of serum tissue transglutaminase (malabsorption or celiac disease)
7. Karyotype (girls) and/or Noonan’s testing
8. Thyroid function tests: thyroxine (T4) and thyroid-stimulating hormone (TSH)
9. IGF-1 (IGFBP-3 is an alternative for children < 4 or in malnourished individuals)
Allen DB, Cuttler L: Clinical practice. Short stature in childhood—challenges and choices. N Engl J Med 2013 Mar 28; 368(13):1220–1228 [PMID: 23534561].
Clayton PE et al: Management of the child born small for gestational age through to adulthood: a consensus statement of the International Societies of Pediatric Endocrinology and the Growth Hormone Research Society. J Clin Endocrinol Metab 2007;92:804 [PMID: 17200164].
Cook DM, Rose SR. A review of guidelines for use of growth hormone in pediatric and transition patients. Pituitary 2012 Sep;15(3):301–310 [PMID: 22271255].
Ozono K et al. Pediatric aspects of skeletal dysplasia. Pediatr Endocrinol Rev 2012 Oct;10(Suppl 1):35–43 [PMID: 23330244].
Rosenbloom AL: Mecasermin (recombinant human insulin-like growth factor 1). Adv Ther 2009;26:40 [PMID: 19198769].
TALL STATURE
Although growth disturbances are usually associated with short stature, potentially serious pathologic conditions may also be associated with tall stature and excessive growth (Table 34–3). Excessive GH secretion is rare and generally associated with a functioning pituitary adenoma. GH excess leads to gigantism if the epiphyses are open and to acromegaly if the epiphyses are closed. The diagnosis is confirmed by finding elevated random GH and IGF-1 levels and failure of GH suppression during an oral glucose tolerance test. Precocious puberty can also cause tall stature for age or rapid growth, but would be associated with early signs of puberty and an advanced bone age. Obese youth are also often tall for age, but do not achieve a taller final height.
Table 34–3. Causes of tall stature.
Because the upper limit of acceptable height in both sexes is increasing, concerns about excessive growth in girls are less frequent than in the past. When such concerns arise, the family history, growth curve, pubertal stage, and assessment of skeletal maturation allow estimation of final adult height. Reassurance, counseling, and education may alleviate family and personal concerns. Rarely, when the predicted height is excessive and felt to be psychologically unacceptable, brief estrogen therapy may be used to accelerate bone maturation and shorten the growth period.
Neylon OM, Werther GA, Sabin MA. Overgrowth syndromes. Curr Opin Pediatr 2012 Aug;24(4):505–511 [PMID: 227059].
DISORDERS OF THE POSTERIOR PITUITARY GLAND
The posterior pituitary (neurohypophysis) is an extension of the ventral hypothalamus. Its two principal hormones—oxytocin and arginine vasopressin—are synthesized in the supraoptic and paraventricular nuclei of the ventral hypothalamus. After synthesis, these peptide hormones are packaged in granules with specific neurophysins and transported via the axons to their storage site in the posterior pituitary. Vasopressin is essential for water balance; it acts primarily on the kidney to promote reabsorption of water from urine. Oxytocin is most active during parturition and breast feeding and is not discussed further here.
ARGININE VASOPRESSIN (ANTIDIURETIC HORMONE) PHYSIOLOGY
Vasopressin release is controlled primarily by serum osmolality and intravascular volume. Release is stimulated by minor increases in plasma osmolality (detected by osmoreceptors in the anterolateral hypothalamus) and large decreases in intravascular volume (detected by baroreceptors in the cardiac atria). Disorders of vasopressin release and action include (1) central (neurogenic) diabetes insipidus, (2) nephrogenic diabetes insipidus (see Chapter 24), and (3) the syndrome of inappropriate secretion of anti-diuretic hormone (see Chapter 45).
CENTRAL DIABETES INSIPIDUS
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Polydipsia, polyuria (> 2 L/m2/d), nocturia, dehydration, and hypernatremia.
Polydipsia, polyuria (> 2 L/m2/d), nocturia, dehydration, and hypernatremia.
 Inability to concentrate urine after fluid restriction (urine specific gravity < 1.010; urine osmolality < 300 mOsm/kg).
Inability to concentrate urine after fluid restriction (urine specific gravity < 1.010; urine osmolality < 300 mOsm/kg).
 Plasma osmolality > 300 mOsm/kg with urine osmolality < 600 mOsm/kg.
Plasma osmolality > 300 mOsm/kg with urine osmolality < 600 mOsm/kg.
 Low plasma vasopressin with antidiuretic response to exogenous vasopressin.
Low plasma vasopressin with antidiuretic response to exogenous vasopressin.
 General Considerations
General Considerations
Central diabetes insipidus (DI) is an inability to synthesize and release vasopressin. Without vasopressin, the kidneys cannot concentrate urine, causing excessive urinary water loss. Genetic causes of central DI are rare and include mutations in the vasopressin gene (mostly in the neurophysin portion of the vasopressin precursor) and the WFS1 gene that causes DI, diabetes mellitus, optic atrophy, and deafness (Wolfram or DIDMOAD syndrome). Transcription factor mutations, such as PROP1 and PIT1, that are known to be associated with other anterior pituitary hormone deficiencies are not typically associated with DI. Midline brain abnormalities, such as septo-optic dysplasia and holoprosencephaly, are also associated with central DI. Traumatic brain injury or neurosurgery in or near the hypothalamus or pituitary can cause transient or permanent DI. Traumatic DI often has three phases. Initially, transient DI is caused by edema in the hypothalamus or pituitary area. In 2–5 days, unregulated release of vasopressin from dying neurons causes the syndrome of inappropriate secretion of antidiuretic hormone (SIADH). Finally, permanent DI occurs if a sufficient number of vasopressin neurons are destroyed.
Tumors and infiltrative diseases of the hypothalamus and pituitary may cause DI. In children with craniopharyngioma, DI usually develops after neurosurgical intervention. In contrast, germinomas often present with DI. Germinomas may be undetectable for several years; consequently, children with unexplained DI should have regularly repeated magnetic resonance imaging (MRI). Infiltrative diseases such as histiocytosis and lymphocytic hypophysitis can cause DI. In these conditions, as in germinomas, MRI scans characteristically show thickening of the pituitary stalk. Infections involving the base of the brain also cause transient DI.
 Clinical Findings
Clinical Findings
Onset of DI is often abrupt, characterized by polyuria, nocturia, enuresis, and intense thirst. Children with DI typically crave cold water. Hypernatremia, hyperosmolality, and dehydration occur if insufficient fluid intake due to lack of access or impaired thirst mechanism does not keep up with urinary losses. In infants, symptoms may also include failure to thrive, vomiting, constipation, and unexplained fevers. Some infants may present with severe dehydration, circulatory collapse, and seizures. Vasopressin deficiency may be masked in patients with panhypopituitarism due to the impaired excretion of free water associated with adrenal insufficiency. Treating these patients with glucocorticoids may unmask their DI.
DI is confirmed when serum hyperosmolality is associated with urine hypoosmolarity. If the history indicates that the child can go through the night comfortably without drinking, outpatient testing is appropriate. Oral fluid intake is prohibited after midnight. Osmolality, sodium, and specific gravity of the first morning void are obtained. If urine specific gravity is greater than 1.015, DI is excluded. If urine is not concentrated, a blood sample is obtained within a few minutes of the urine collection for osmolality, sodium, creatinine, and calcium concentration.
If screening results are unclear or if symptoms preclude safely withholding fluids at home, a water deprivation test performed in the hospital is indicated. In this test, fluid is withheld and the child is monitored. Serum osmolality greater than 290 mOsm/kg associated with inappropriately dilute urine (osmolality less than 600 mOsm/kg) is diagnostic for DI. Low serum vasopressin concentration and an antidiuretic response to vasopressin administration at the end of the test distinguishes central from nephrogenic DI. Children with central DI should have a head MRI scan with contrast to look for tumors or infiltrative processes. The posterior pituitary “bright spot” on MRI is often absent in DI.
Primary polydipsia must be distinguished from DI. Children with primary polydipsia tend to have lower serum sodium levels and usually can concentrate their urine with overnight fluid deprivation. Some may have secondary nephrogenic DI due to dilution of the renal medullary interstitium and decreased renal concentrating ability, but this resolves with restriction of fluid intake.
 Treatment
Treatment
Central DI is treated with oral or intranasal desmopressin acetate (DDAVP). The aim of therapy is to provide antidiuresis that allows uninterrupted sleep and approximately 1 hour of diuresis before the next dose. It is important to note that postsurgical DI can be associated with disruption of thirst mechanism and, for these patients, a prescribed volume of fluid intake needs to be determined. Children hospitalized with acute-onset DI can be managed with intravenous vasopressin. Due to the amount of antidiuresis, intravenous fluids will need to be restricted to two-thirds the maintenance rate and electrolytes closely monitored to avoid water intoxication. Infants with DI should not be treated with DDAVP. Treatment with DDAVP in association with the volume of formula or breast milk needed to ensure adequate caloric intake could cause water intoxication. For this reason, infants are treated with extra free water, rather than DDAVP, to maintain normal hydration. A formula with a low renal solute load and chlorothiazides may be helpful in infants with central DI.
Cheetham T, Baylis PH: Diabetes insipidus in children: pathophysiology, diagnosis and management. Paediatr Drugs 2002;4:785 [PMID: 12431131].
Di Iorgi N et al: Diabetes insipidus—diagnosis and management. Horm Res Paediatr 2012;77:69 [PMID: 22433947].
Rivkees SA et al: The management of central diabetes insipidus in infancy: desmopressin, low renal solute load formula, thiazide diuretics. J Pediatr Endocrinol Metab 2007;20:459 [PMID: 17550208].
Wise-Faberowski L et al: Perioperative management of diabetes insipidus in children. J Neurosurg Anesthesiol 2004;16:14 [PMID: 14676564].
THYROID GLAND
FETAL DEVELOPMENT OF THE THYROID
The fetal thyroid synthesizes thyroid hormone as early as the 10th week of gestation. Thyroid hormone appears in the fetal serum by the 11th week of gestation and progressively increases throughout gestation. The fetal pituitary-thyroid axis functions largely independently of the maternal pituitary-thyroid axis because maternal TSH cannot cross the placenta. However, maternal thyroid hormone can cross the placenta in limited amounts.
At birth, there is a TSH surge peaking at about 70 mU/L within 30–60 minutes. Thyroid hormone serum level increases rapidly during the first days of life in response to this TSH surge. The TSH level decreases to childhood levels within a few weeks. The physiologic neonatal TSH surge can produce a false-positive newborn screen for hypothyroidism (ie, high TSH) if the blood sample for the screen is collected on the first day of life.
PHYSIOLOGY
Hypothalamic thyrotropin-releasing hormone (TRH) stimulates the anterior pituitary gland to release TSH. In turn, TSH stimulates the thyroid gland to take up iodine, and to synthesize and release the active hormones, thyroxine (T4) and triiodothyronine (T3). This process is regulated by negative feedback involving the hypothalamus, pituitary, and thyroid (see Figure 34–1).
T4 is the predominant thyroid hormone secreted by the thyroid gland. Most circulating T3 and T4 are bound to thyroxine-binding globulin (TBG), albumin, and prealbumin. Less than 1% of T3 and T4 exist as free T3 (FT3) and free T4 (FT4). T4 is deiodinated in the tissues to either T3 (active) or reverse T3 (inactive). In peripheral tissues, T3 binds to high-affinity nuclear thyroid hormone receptors in the cytoplasm and translocates to the nucleus, exerting its biologic effects by modifying gene expression.
The T4 level is low in hypothyroidism. It may also be low in premature infants, malnutrition, severe illness, and following therapy with T3. It is not clear whether premature infants with low T4 benefit from treatment. Long-term studies have been proposed to assess cognitive outcomes for high-risk patients.
Total T4 is also low in situations that decrease TBG. TBG levels are decreased in familial TBG deficiency, nephrosis, and in patients receiving androgens. In sepsis, TBG cleavage is increased. Treatment with certain medications (heparin, furosemide, salicylates, and phenytoin) results in abnormal binding to TBG. However, since these effects involve primarily TBG levels, and not thyroid function per se, TSH and FT4 levels remain in the normal range. Conversely, Total T3 and T4 levels may be elevated in conditions associated with increased TBG levels (congenital TBG excess, pregnancy, estrogen therapy) and increased thyroid hormone binding to transport proteins. However, free hormone levels are not affected and patients are euthyroid.
HYPOTHYROIDISM (CONGENITAL & ACQUIRED)
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Growth retardation, decreased physical activity, weight gain, constipation, dry skin, cold intolerance, and delayed puberty.
Growth retardation, decreased physical activity, weight gain, constipation, dry skin, cold intolerance, and delayed puberty.
 Neonates with congenital hypothyroidism often look normal but may have thick tongue, large fontanels, poor muscle tone, hoarseness, umbilical hernia, jaundice, and intellectual retardation.
Neonates with congenital hypothyroidism often look normal but may have thick tongue, large fontanels, poor muscle tone, hoarseness, umbilical hernia, jaundice, and intellectual retardation.
 T4, FT4, and T3 resin uptake are low; TSH levels are elevated in primary hypothyroidism.
T4, FT4, and T3 resin uptake are low; TSH levels are elevated in primary hypothyroidism.
 General Considerations
General Considerations
Thyroid hormone deficiency may be congenital or acquired (Table 34–4). It can be due to defects in the thyroid gland (primary hypothyroidism) or in the hypothalamus or pituitary (central hypothyroidism).

Congenital hypothyroidism occurs in about 1:3000–1:4000 infants. Untreated, it causes severe neurocognitive impairment. Most cases are sporadic resulting from hypoplasia or aplasia of the thyroid gland or failure of the gland to migrate to its normal anatomic location (ie, lingual or sublingual thyroid gland). Another cause of congenital hypothyroidism is dyshormonogenesis due to enzymatic defects in thyroid hormone synthesis. Since antithyroid drugs, including propylthiouracil (PTU) and methimazole, freely cross the placenta, goitrous hypothyroid newborns may be born to hyperthyroid mothers treated with these drugs during pregnancy.
Low T4 levels may also be caused by decreased TSH secretion associated with prolonged glucocorticoid use, dopamine, or somatostatin. Cabbage, soybeans, aminosalicylic acid, thiourea derivatives, resorcinol, phenylbutazone, cobalt, and excessive iodine intake can cause goiter and hypothyroidism during pregnancy. Many of these agents cross the placenta and should be used with caution during pregnancy. Iodine deficiency also causes hypothyroidism. In severe maternal iodine deficiency, both the fetus and the mother are T4-deficient, with irreversible brain damage in the fetus.
Juvenile hypothyroidism, particularly if goiter is present, is usually a result of chronic lymphocytic (Hashimoto) thyroiditis.
Several hundred patients with resistance to thyroid hormone have been described and present with elevations in T4 and/or FT4, with normal TSH. There is often a family history of the disorder. Clinical manifestations are highly variable due to differential expression of thyroid hormone receptor isoforms in different tissues
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Even when the thyroid gland is completely absent, most newborns with congenital hypothyroidism appear normal at birth and gain weight normally for the first few months of life without treatment. Since congenital hypothyroidism must be treated as early as possible to prevent intellectual impairment, the diagnosis should be based on the newborn screening test and not on signs or symptoms. Jaundice associated with an unconjugated hyperbilirubinemia may be present in newborns with congenital hypothyroidism. Some infants may have obvious findings of thick tongue, hypotonia, large fontanelles, constipation, umbilical hernia, hoarse cry, and dry skin.
Juvenile hypothyroidism often presents with short stature and abnormal weight gain. Other findings include delayed epiphyseal development, delayed closure of fontanels, and retarded dental eruption. The skin may be dry, thick, scaly, coarse, pale, cool, or mottled, or have a yellowish tinge. The hair may be dry, coarse, or brittle. Lateral thinning of the eyebrows may occur. Musculoskeletal findings include hypotonia and a slow relaxation component of deep tendon reflexes (best appreciated in the ankles). Muscular hypertrophy (Kocher-Debré-Semélaigne syndrome) is not commonly seen in congenital hypothyroidism. Other findings include physical and mental sluggishness, nonpitting myxedema, constipation, large tongue, hypothermia, bradycardia, hoarse voice or cry, umbilical hernia, and transient deafness. Puberty may be delayed. Metromenorrhagia may occur in older girls. Sometimes, hypothyroidism induces pseudopuberty. Galactorrhea can also occur, due to stimulation of prolactin secretion.
In hypothyroidism resulting from enzymatic defects, ingestion of goitrogens, or chronic lymphocytic thyroiditis, the thyroid gland may be enlarged. Thyroid enlargement in children is usually symmetrical, and the gland is moderately firm and not nodular. In chronic lymphocytic thyroiditis, however, the thyroid frequently has a cobblestone surface.
B. Laboratory Findings
Total T4 and FT4 levels are decreased. T3 resin uptake (T3RU) is low. In primary hypothyroidism, the serum TSH level is elevated. In central hypothyroidism, the TSH level may be low or inappropriately normal. Circulating autoantibodies to thyroid peroxidase and thyroglobulin may be present. Serum prolactin may be elevated, resulting in galactorrhea. Serum GH may be decreased, with subnormal GH response to stimulation in children with severe primary hypothyroidism, as well as low IGF-1 or IGFBP-3 levels, or both.
C. Imaging
Thyroid imaging, while helpful in establishing the cause of congenital hypothyroidism, does not affect the treatment plan and is not necessary. Bone age is delayed. Centers of ossification, especially of the hip, may show multiple small centers or a single stippled, porous, or fragmented center (epiphyseal dysgenesis). Cardiomegaly is common. Longstanding primary hypothyroidism may be associated with thyrotrophic hyperplasia characterized by an enlarged sella or pituitary gland.
D. Screening Programs for Neonatal Hypothyroidism
All newborns should be screened for congenital hypothyroidism shortly after birth as most do not have suggestive physical findings. Depending on the state, the newborn screen measures either the total T4 or TSH level. Abnormal newborn screening results should be confirmed immediately with a T4 and TSH level. Treatment should be started as soon as possible. Initiation of treatment in the first month of life and good compliance during infancy usually results in a normal neurocognitive outcome.
E. Conditions Associated With Hypothyroidism
Children with Down syndrome, Turner syndrome, and autoimmune diseases such as celiac disease, vitiligo, alopecia, and type 1 diabetes are at an increased risk for the development of acquired autoimmune hypothyroidism. A detailed family history may reveal the presence of multiple autoimmune diseases in the family members of the affected individual. Individuals at a high risk based on a chromosomal disorder or other autoimmune disease benefit from careful monitoring of growth and development, routine screening (in the case of Down syndrome, Turner syndrome, and type 1 diabetes), and a low threshold for measurement of thyroid function.
Central hypothyroidism is associated with other disorders of the hypothalamus and pituitary including congenital defects such as septo-optic dysplasia and acquired defects such as tumors in the hypothalamic/pituitary region.
 Treatment
Treatment
Levothyroxine (75–100 mcg/m2/d) is the drug of choice for acquired hypothyroidism. In neonates with congenital hypothyroidism, the initial dose is 10–15 mcg/kg/d. Serum total T4 or FT4 concentrations are used to monitor the adequacy of initial therapy because the normally high neonatal TSH may not normalize for several days to weeks. Subsequently, T4 and TSH are used in combination, as elevations of serum TSH are sensitive early indicators of the need for increased medication or better compliance.
Delahunty C et al: Levels of neonatal thyroid hormone in preterm infants and neurodevelopmental outcomes at 5½ years: millennium cohort study. J Clin Endocrinol Metabl 2010;95: 4898–4908 [PMID: 20719832].
LaFranchi S: Approach to the diagnosis and treatment of congenital hypothyroidism. J Clin Endocrinol Metab 2011;96: 2959–2967 [PMID: 21976744].
Schmaltz C: Thyroid hormones in the neonate: an overview of physiology and clinical correlation. Adv Neonatal Care 2012;12:217–222 [PMID: 22864001].
van der Sluijs Veer L et al: Evaluation of cognitive and motor development in toddlers with congenital hypothyroidism diagnosed by neonatal screening. J Dev Behav Pediatr 2012;33: 633–640 [PMID: 23027136].
THYROIDITIS
1. Chronic Lymphocytic Thyroiditis (Chronic Autoimmune Thyroiditis, Hashimoto Thyroiditis)
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Firm, freely movable, nontender, diffusely enlarged thyroid gland.
Firm, freely movable, nontender, diffusely enlarged thyroid gland.
 Thyroid function is usually normal but may be elevated or decreased depending on the stage of the disease.
Thyroid function is usually normal but may be elevated or decreased depending on the stage of the disease.
 General Considerations
General Considerations
Chronic lymphocytic thyroiditis is the most common cause of goiter and acquired hypothyroidism in childhood. It is more common in girls, and the incidence peaks during puberty. The disease is caused by an autoimmune attack on the thyroid. Increased risk of thyroid autoimmunity (and other endocrine autoimmune disorders) is associated with certain histocompatibility alleles.
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
The thyroid is characteristically enlarged, firm, freely movable, nontender, and symmetrical. It may be nodular. Onset is usually insidious. Occasionally patients note a sensation of tracheal compression or fullness, hoarseness, and dysphagia. No local signs of inflammation or systemic infection are present. Most patients are euthyroid. Some patients are symptomatically hypothyroid, and few patients are symptomatically hyperthyroid.
B. Laboratory Findings
Laboratory findings vary. Serum concentrations of TSH, T4, and FT4 are usually normal. Some patients are hypothyroid with an elevated TSH and low thyroid hormone levels. A few patients are hyperthyroid with a suppressed TSH and elevated thyroid hormone levels. Thyroid antibodies (antithyroglobulin, antithyroid peroxidase) are frequently elevated. Thyroid uptake scan adds little to the diagnosis. Surgical or needle biopsy is diagnostic but seldom necessary.
 Treatment
Treatment
There is controversy about the need to treat chronic lymphocytic thyroiditis with normal thyroid function. Full replacement doses of thyroid hormone may decrease the size of the thyroid, but may also result in hyperthyroidism. Hypothyroidism commonly develops over time. Consequently, patients require lifelong surveillance. Children with documented hypothyroidism should receive thyroid hormone replacement.
2. Acute (Suppurative) Thyroiditis
Acute thyroiditis is rare. The most common causes are group A streptococci, pneumococci, Staphylococcus aureus, and anaerobes. Oropharyngeal organisms are thought to reach the thyroid via a patent foramen cecum and thyroglossal duct remnant. Thyroid abscesses may form. The patient is toxic with fever and chills. The thyroid gland is enlarged and exquisitely tender with associated erythema, hoarseness, and dysphagia. Thyroid function tests are typically normal. Patients have a leukocytosis, “left shift,” and elevated erythrocyte sedimentation rate. Specific antibiotic therapy should be administered.
3. Subacute (Nonsuppurative) Thyroiditis
Subacute thyroiditis (de Quervain thyroiditis) is rare. It is thought to be caused by viral infection with mumps, influenza, echovirus, coxsackievirus, Epstein-Barr virus, or adenovirus. Presenting features are similar to acute thyroiditis—fever, malaise, sore throat, dysphagia, and thyroid pain that may radiate to the ear. The thyroid is firm and enlarged. Sedimentation rate is elevated. In contrast to acute thyroiditis, the onset is generally insidious and serum thyroid hormone concentrations may be elevated.
HYPERTHYROIDISM
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Nervousness, emotional lability, hyperactivity, fatigue, tremor, palpitations, excessive appetite, weight loss, increased perspiration, and heat intolerance.
Nervousness, emotional lability, hyperactivity, fatigue, tremor, palpitations, excessive appetite, weight loss, increased perspiration, and heat intolerance.
 Goiter, exophthalmos, tachycardia, widened pulse pressure, systolic hypertension, weakness, and smooth, moist, warm skin.
Goiter, exophthalmos, tachycardia, widened pulse pressure, systolic hypertension, weakness, and smooth, moist, warm skin.
 TSH is suppressed. Thyroid hormone levels (T4, FT4, T3, T3RU) are elevated.
TSH is suppressed. Thyroid hormone levels (T4, FT4, T3, T3RU) are elevated.
 General Considerations
General Considerations
In children, most cases of hyperthyroidism are due to Graves disease, caused by antibodies directed at the TSH receptor that stimulate thyroid hormone production. Hyperthyroidism may also be due to acute, subacute, or chronic thyroiditis; autonomous functioning thyroid nodules; tumors producing TSH; McCune-Albright syndrome; exogenous thyroid hormone excess; and acute iodine exposure.
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Hyperthyroidism is more common in females than males. In children, it most frequently occurs during adolescence. The course of hyperthyroidism tends to be cyclic, with spontaneous remissions and exacerbations. Symptoms include worsening school performance, poor concentration, fatigue, hyperactivity, emotional lability, nervousness, personality disturbance, insomnia, weight loss (despite increased appetite), palpitations, heat intolerance, increased perspiration, diarrhea, polyuria, and irregular menses. Signs include tachycardia, systolic hypertension, increased pulse pressure, tremor, proximal muscle weakness, and moist, warm, skin. Accelerated growth and development may occur. Thyroid storm is a rare condition characterized by fever, cardiac failure, emesis, delirium, coma, and death. Most cases of Graves disease are associated with a diffuse firm goiter. A thyroid bruit and thrill may be present. Many cases are associated with exophthalmos, but severe ophthalmopathy is rare.
B. Laboratory Findings
TSH is suppressed. T4, FT4, T3, and FT3 are elevated except in rare cases in which only the serum T3 is elevated (T3 thyrotoxicosis). The presence of thyroid-stimulating immunoglobulin (TSI) confirms the diagnosis of Graves disease. TSH receptor–binding antibodies (TRaB) are usually elevated.
C. Imaging
In Graves disease, radioactive iodine uptake by the thyroid is increased, whereas in subacute and chronic thyroiditis it is decreased. An autonomous hyperfunctioning nodule takes up iodine and appears as a “hot nodule” while the surrounding tissue has decreased iodine uptake. In children with hyperthyroidism, bone age may be advanced. In infants, accelerated skeletal maturation may be associated with premature fusion of the cranial sutures. Long-standing hyperthyroidism causes osteoporosis.
 Differential Diagnosis
Differential Diagnosis
Hypermetabolic states (severe anemia, chronic infections, pheochromocytoma, and muscle-wasting disease) may resemble hyperthyroidism clinically but differ in thyroid function tests.
 Treatment
Treatment
A. General Measures
In untreated hyperthyroidism, strenuous physical activity should be avoided. Bed rest may be required in severe cases.
B. Medical Treatment
1. β-Adrenergic blocking agents— These agents are adjuncts to therapy. They can rapidly ameliorate symptoms such as nervousness, tremor, and palpitations, and are indicated in severe disease with tachycardia and hypertension. β1-Specific agents such as atenolol are preferred because they are more cardioselective.
2. Antithyroid agents (propylthiouracil and methimazole)— Antithyroid agents are frequently used in the initial treatment of childhood hyperthyroidism. These drugs interfere with thyroid hormone synthesis, and usually take a few weeks to produce a clinical response. Adequate control is usually achieved within a few months. If medical therapy is unsuccessful, more definitive therapy, such as radioablation of the thyroid or thyroidectomy, should be considered.
In response to reports of severe hepatotoxicity related to PTU, recent recommendations state that PTU should not be used in infants, children, or adolescents, except when methimazole is contraindicated due to hypersensitivity or pregnancy.
A. INITIAL DOSAGE— Methimazole is initiated at a dose of 10–60 mg/d (0.5–1 mg/kg/d) given once a day. Initial dosing is continued until FT4 or T4 have normalized and signs and symptoms have subsided.
B. MAINTENANCE— The optimal dose of antithyroid agent for maintenance treatment remains unclear. Recent studies suggest that 10–15 mg/d of methimazole provides adequate long-term control in most patients with a minimum of side effects. If the TSH becomes elevated, many providers decrease the dose of the antithyroid agent. Some providers continue the same dose of antithyroid agent and add exogenous thyroid hormone replacement. Treatment usually continues for 2 years with the goal of inducing remission. If thyroid hormone levels are stable, a trial off medication could be considered at that point.
C. TOXICITY— If rash, vasculitis, arthralgia, arthritis, granulocytopenia, or hepatitis occur, the drug must be discontinued.
3. IODIDE— Large doses of iodide usually produce a rapid but short-lived blockade of thyroid hormone synthesis and release. This approach is recommended only for acute management of severely thyrotoxic patients.
C. Radiation Therapy
Radioactive iodine ablation of the thyroid is usually reserved for children with Graves disease who do not respond to antithyroid agents, develop adverse effects from the antithyroid agents, fail to achieve remission after several years of medical therapy, or have poor medication adherence. With recent concerns regarding potential hepatotoxicity of antithyroid medications, some pediatric endocrinologists advocate radioablation as first-line therapy for children with Graves disease. Antithyroid agents should be discontinued 4–7 days prior to radioablation to allow radioiodine uptake by the thyroid.131 I is administered orally which concentrates in the thyroid and results in gradual ablation of the gland. In the first 2 weeks following radioablation, hyperthyroidism may worsen as thyroid tissue is destroyed and thyroid hormone is released. Therapy with a β-adrenergic antagonist may be necessary for a few months until FT4 and T4 fall into the normal range. In most cases, hypothyroidism develops and thyroid hormone replacement is needed. Long-term follow-up studies have not shown any increased incidence of thyroid cancer, leukemia, infertility, or birth defects when ablative doses of 131 I were used.
D. Surgical Treatment
Subtotal and total thyroidectomy are infrequently used in children with Graves disease. Surgery is indicated for extremely large goiters, goiters with a suspicious nodule, very young or pregnant patients, or patients refusing radioiodine ablation. Before surgery, a β-adrenergic blocking agent is given to treat symptoms, and antithyroid agents are given for several weeks to minimize the surgical risks associated with hyperthyroidism. Iodide (eg, Lugol solution, 1 drop every 8 hours, or saturated solution of potassium iodide, 1–2 drops daily) is given for 1–2 weeks prior to surgery to reduce thyroid vascularity and inhibit release of thyroid hormone. Surgical complications include hypoparathyroidism, recurrent laryngeal nerve damage, and rarely, death. An experienced thyroid surgeon is crucial to good surgical outcome. After thyroidectomy, patients become hypothyroid and need thyroid hormone replacement.
 Course & Prognosis
Course & Prognosis
Partial remissions and exacerbations may continue for several years. Treatment with an antithyroid agent results in prolonged remissions in one-third to two-thirds of children.
Bauer AJ: Approach to the pediatric patient with Graves’ disease: when is definitive therapy warranted? J Clin Endocrinol Metab 2011;96:580–588 [PMID: 21378220].
Lee JA et al: The optimal treatment for pediatric Graves’ disease is surgery. J Clin Endocrinol Metab 2007;92:80 [PMID: 17341575].
Rivkees SA, Dinauer C: An optimal treatment for pediatric Graves’ disease is radioiodine. J Clin Endocrinol Metab 2007;92:797 [PMID: 17341574].
Yuan L, Yang J: Radioiodine treatment in pediatric Graves’ disease and thyroid carcinoma. J Pediatr Endocrinol Metab 2011;24:877–883 [PMID: 22308835].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


