Endocrine Causes of Hypoglycemia
Andrea Kelly and Charles A. Stanley
Hypoglycemic disorders are generally classified as either fasting hypoglycemia or reactive or postprandial hypoglycemia. The risk of hypoglycemia varies depending upon the underlying disorder. With the exception of late dumping syndrome, a form of reactive hypoglycemia associated with Nissen fundoplication or a gastric tube, childhood hypoglycemia is uniformly a disorder of fasting adaptation. Specific endocrine disorders and associated risk factors for hypoglycemia are discussed in this chapter. Defects in the metabolic systems are discussed in Chapter 134.
FASTING ADAPTATION
Normal fasting adaptation involves 5 major systems: 4 metabolic systems (hepatic gluconeogenesis, hepatic glycogenolysis, adipose tissue lipolysis, and oxidation of fatty acids for hepatic ketogenesis), as well as the hormonal system that regulates these metabolic systems. Within 2 to 3 hours of a meal, when intestinal absorption of glucose ceases, hepatic glycogenolysis and gluconeogenesis produce glucose to meet the requirement for brain glucose oxidation and to prevent a decline in blood glucose concentrations. Prolonged fasting of 8 to 12 hours or more depletes glucose and glycogen stores, and adipose tissue lipolysis is activated to provide fatty acids used by muscle and for ketogenesis by the liver. In young children fatty acids become the main fuel source for most of the body after 12 to 24 hours of fasting. Glucose is spared for use by the brain. Ketones become a major fuel for the brain to further spare glucose utilization. The changing serum levels of these fuels obtained during fasting reflect these metabolic processes as shown in Figure 545-1.
The metabolic systems for fasting adaptation are subject to strict hormonal regulation. Insulin negatively regulates all 4 fasting metabolic systems. As blood glucose concentrations decline during the initial phase of fasting, insulin levels fall. This allows increased rates of glucose release from hepatic glycogenolysis and gluconeogenesis. As fasting progresses, the further fall in blood glucose and further suppression of insulin release permit activation of lipolysis and fatty acid oxidation.
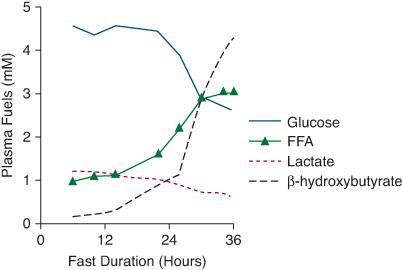
FIGURE 545-1. Normal changes in metabolic fuels during fasting. Levels of these fuels obtained during the fast and at the time of hypoglycemia reflect the operation of the metabolic systems. Within 2 to 3 hours of a meal, hepatic gluconeogenesis and glycogenolysis are activated to maintain blood glucose. As hepatic glycogen stores are depleted and gluconeogenesis is activated, blood glucose and lactate levels decline. With more prolonged fasting, adipose tissue lipolysis is activated and plasma free fatty acid concentrations increase, followed by a dramatic increase in β-hydroxybutyrate as the fatty acids are oxidized to ketones in the liver. The high levels of ketones can be used by the brain to spare glucose utilization.
The inhibitory effects of insulin is counterbalanced by actions of glucagon, epinephrine, cortisol, and growth hormone. These hormones activate the metabolic systems required for fasting adaptation. Glucagon release, stimulated by decreasing plasma glucose and insulin, acutely stimulates hepatic glycogenolysis and promotes hepatic gluconeogenesis and ketogenesis. Epinephrine acutely stimulates glycogenolysis and gluconeogenesis but also is a potent stimulator of adipose tissue lipolysis, thereby furnishing free fatty acids for hepatic ketogenesis. Less acute changes result from release of cortisol and growth hormone. Cortisol inhibits peripheral tissue glucose uptake and stimulates hepatic gluconeogenesis. Growth hormone also inhibits glucose utilization and has important effects on stimulating adipose tissue lipolysis.
The rate of progression through the phases of fasting adaptation is largely determined by the ratio of brain versus body weight. Therefore, fasting tolerance is lower in infants and children than in adults, with fasting hyperketonemia occurring in 18 hours in normal neonates, 24 hours in older infants, and 36 hours in children over 1 year.
DIAGNOSIS OF HYPOGLYCEMIA
Symptoms of hypoglycemia are due to decreased metabolic substrate for the brain and elevated catecholamines. They include shakiness, dizziness, sweating, pallor, sudden moodiness, headache, irritability or lethargy (especially in the infant), inattention, and sometimes tingling or even seizures.
A plasma glucose level below 50 mg/dL is commonly used as a diagnostic threshold for diagnosis of hypoglycemia. A treatment goal for a child with a hypoglycemic disorder is to maintain blood glucose concentrations above 70 mg/dL. This therapeutic target of more than 70 mg/dL is particularly important in children with hypoglycemia due to hyperinsulinism, since these infants are unable to produce ketones as an alternative fuel for the brain when glucose is low.
THE CRITICAL SAMPLE AND FASTING TEST
Laboratory specimens obtained at the time of hypoglycemia (blood glucose < 50 mg/dL) are referred to as the critical samples. During hypoglycemia, fasting compensatory mechanisms become manifest. Deviations from the expected pattern of plasma fuels indicate a specific site of defect in one of the systems of fasting adaptation.
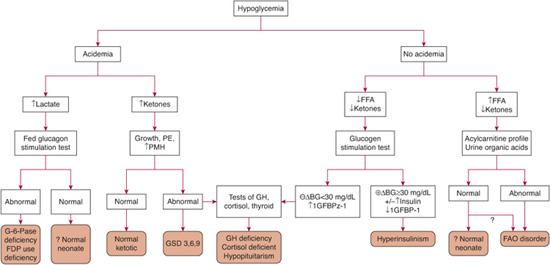
The most useful tests to obtain at the time of hypoglycemia are the plasma bicarbonate and urinary ketones because the presence or absence of these form the basis for the initial differential diagnosis as outlined in Figure 545-2. Acidemia may arise from ketosis or lactic acidosis. Ketosis suggests normal fasting adaptation, normal but shortened fasting adaptation (ketotic hypoglycemia), a defect in glycogenolysis, or a defect in counterregulatory hormone production. Lactic acidosis suggests a defect in gluconeogenesis. In contrast, the absence of acidemia suggests either hyperinsulinism or a fatty acid oxidation disorder. Both are associated with inappropriately low or absent urinary ketones but can be differentiated by plasma free fatty acid concentrations. With hyperinsulinism, lipolysis is suppressed and plasma free fatty acid concentrations are low. With a defect in fatty acid oxidation, lipolysis is activated normally and free fatty acid levels are increased. Thus, obtaining a blood sample for free fatty acids, b-hydroxybutyrate, and lactate at the time of hypoglycemia will provide valuable information on the likely site of the defect in fasting adaptation. The neonate with hypoglycemia due to pituitary deficiencies can have biochemical findings that resemble hyperinsulinism.
Measurements of the important hormonal regulators of fasting adaptation in the critical sample are also recommended. In children with hypoglycemia due to hyperinsulinism, insulin levels are often not dramatically elevated at the time of hypoglycemia (eFig. 545.1  ). For this specific reason, the glucagon stimulation test was developed. In the normal child, hepatic glycogen stores are depleted during fasting by the time hypoglycemia occurs. Administration of glucagon does not increase blood glucose. In contrast, in the child with hyperinsulinism, glycogenolysis is suppressed so that glycogen stores are inappropriately not depleted. Glucagon administration leads to a robust glycemic response (Δ blood glucose > 30 mg/dL).
). For this specific reason, the glucagon stimulation test was developed. In the normal child, hepatic glycogen stores are depleted during fasting by the time hypoglycemia occurs. Administration of glucagon does not increase blood glucose. In contrast, in the child with hyperinsulinism, glycogenolysis is suppressed so that glycogen stores are inappropriately not depleted. Glucagon administration leads to a robust glycemic response (Δ blood glucose > 30 mg/dL).
Growth hormone and cortisol levels at the time of hypoglycemia may not provide reliable diagnostic information. However, consideration of hypopituitarism is important in the infant with hypoglycemia. The approach to diagnosis of these disorders is discussed in Chapters 521 and 523. Additional critical sample studies include an acylcarnitine profile, total and free carnitine, and urine organic acids, all of which provide clues to the diagnoses of specific defects in fatty acid oxidation, as discussed in Chapter 150.
HYPERINSULINISM
Insulin secretion is normally triggered when glucose levels exceed 70 to 90 mg/dL, being controlled by a “thermostat-like” mechanism set by islet glucokinase. Hyperinsulinism refers to the group of hypoglycemic disorders that arise from dysregulated insulin secretion. In infants, hypoglycemia due to dysregulated insulin secretion can be a temporary issue, as occurs in the infant of the diabetic mother (discussed in Chapter 48) and in perinatal stress-induced hyperinsulinism. Alternatively, hyperinsulinism may arise from genetic defects in the pathways of insulin secretion. In the older child, acquired hyperinsulinism as a result of insulinoma, antibody-mediated processes, or medication, are considerations, but congenital defects remain a possibility. The hypoglycemia is frequently severe, and affected infants and children may require far more than the normal glucose infusion rate (6–8 mg/kg/min) to maintain blood glucose in the normal range (> 30–35 mg/kg/min).1-8
 PERINATAL STRESS-INDUCED HYPERINSULINISM
PERINATAL STRESS-INDUCED HYPERINSULINISM
Persistent hypoglycemia is common in infants born small for gestational age, to mothers with preeclampsia, or in the setting of other peripartum stress. Hyperinsulinism is now recognized as the cause of this persistent hypoglycemia,11,12 but the mechanism remains poorly understood.13 Perinatal stress-induced hyperinsulinism can last for a few days to several weeks, but may persist for 2 to 6 months.13
Perinatal stress-induced hyperinsulinism is generally amenable to treatment with diazoxide. Fluid retention due to diazoxide can be a limiting factor in its use in infants with significant prematurity and lung disease. In such infants, hydro-chlorothiazide or other diuretics are empirically initiated and signs of fluid retention monitored. If diazoxide is ineffective, octreotide or continuous feeds are an alternative therapy.
 CONGENITAL HYPERINSULINISM
CONGENITAL HYPERINSULINISM
Congenital hyperinsulinism is the most common cause of recurrent hypoglycemia in the newborn. Our current understanding of glucose-stimulated insulin secretion is shown in Figure 545-3. 
KATP Hyperinsulinism
Mutations in the genes encoding sulfonylurea receptor-1 and Kir6.2, which compose the β-cell KATP channel, are responsible for the most common form of congenital hyperinsulinism, KATP hyperinsulinism.21 Recessive and dominant22,23 forms of KATP hyperinsulinism result in dysfunction of all of the pancreatic islet cells (eFig. 545.2  ). The incidence of KATP hyperinsulinism is estimated to be 1/20,00 to 1/40,000 live births, with an increased incidence in Saudi Arabia, the Ashkenazi-Jewish population, and a subset of the Finnish population.24-27
). The incidence of KATP hyperinsulinism is estimated to be 1/20,00 to 1/40,000 live births, with an increased incidence in Saudi Arabia, the Ashkenazi-Jewish population, and a subset of the Finnish population.24-27
Infants with KATP hyperinsulinism are generally born large for gestational age, and hypoglycemia is evident within the first few days of life. The recessive and focal forms of KATP hyperinsulinism are usually resistant to medical therapy with diazoxide, whereas dominant KATP hyperinsulinism is diazoxide responsive. Octreotide is the second-line drug for diazoxide-unresponsive patients, but is often not effective for chronic treatment because of tachyphylaxis. Pancreatectomy is recommended for infants with KATP hyperinsulinism in whom blood glucose cannot safely be maintained above 70 mg/dL.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


