Duodenal and Intestinal Atresia and Stenosis
Congenital intestinal obstruction occurs in approximately 1 : 2000 live births and is a common cause of admission to a neonatal surgical unit, accounting for up to one-third of all admissions.1 Morphologically, congenital defects related to continuity of the intestine can be divided into either atresia or stenosis. Together, they constitute one of the most common etiologies of neonatal intestinal obstruction.2–4 See Chapter 29 for information about pyloric atresia.
Duodenal Atresia and Stenosis
Congenital duodenal atresia and stenosis is a frequent cause of intestinal obstruction and occurs in 1 per 5000 to 10,000 live births, affecting boys more commonly than girls.5 More than 50% of affected patients have associated congenital anomalies, with trisomy 21 occurring in approximately 30% of patients.6,7 Operative correction is accomplished via a duodenoduodenostomy, with or without tapering duodenoplasty. This can be performed either laparoscopically or open. Early postoperative survival rates of greater than 90% should be expected.7–11
Etiology
Congenital duodenal obstruction can occur due to an intrinsic or extrinsic lesion.12 The most common cause of duodenal obstruction is atresia.7 This intrinsic lesion is most commonly believed to be caused by a failure of recanalization of the fetal duodenum resulting in complete obstruction. Early in the fourth week of gestation, the duodenum begins to develop from the distal foregut and the proximal midgut. During the fifth and sixth weeks of gestation, the duodenal lumen temporarily obliterates due to proliferation of its epithelial cells. Vacuolation due to degeneration of the epithelial cells during the 11th week of gestation then leads to duodenal recanalization.13 An embryologic insult during this period can lead to an intrinsic web, atresia, or stenosis. The extrinsic form of duodenal obstruction is due to defects in the development of neighboring structures such as the pancreas, a preduodenal portal vein, or malrotation and Ladd’s bands.14,15
Annular pancreas as an etiology for duodenal obstruction warrants special mention as this form of obstruction is likely due to failure of duodenal development rather than a true constricting lesion. Thus, the presence of an annular pancreas is simply a visible indication of an underlying atresia or stenosis.16 Between the fourth and eighth week of gestation, the pancreatic buds merge. In annular pancreas, the tip of the ventral pancreas becomes fixed to the duodenal wall forming a nondistensible, ring-like or annular portion of pancreatic tissue surrounding the descending part of the duodenum.13 In annular pancreas associated with duodenal obstruction, the distal biliary tree is often abnormal and may open proximal or distal to the atresia or stenosis.17,18 Other reported biliary abnormalities associated with duodenal obstruction include biliary atresia, gallbladder agenesis, stenosis of the common bile duct, choledochal cyst, and immune deficiency.19–24
Classification
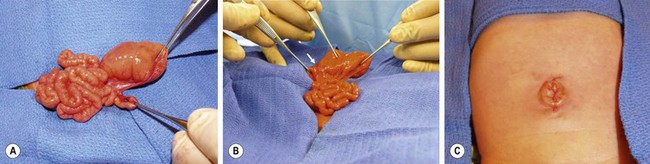
Anatomically, duodenal obstructions are classified as either atresias or stenoses. An incomplete obstruction, due to a fenestrated web or diaphragm, is considered a stenosis. Most stenoses involve the third and/or fourth part of the duodenum. Atresias, or complete obstruction, are further classified into three morphologic types (Fig. 30-1). Type I atresias account for more than 90% of all duodenal obstructions and contain a lumenal diaphragm that includes mucosal and submucosal layers. A diaphragm that has ballooned distally (windsock) is a type I atresia.25,26 It is important to understand that the anatomy of the windsock may lead to a portion of the dilated duodenum actually being distal to the actual obstruction (Fig. 30-2). Type II atresias are characterized by a dilated proximal and collapsed distal segment connected by a fibrous cord. Type III atresias have an obvious gap separating the proximal and distal duodenal segments.27

FIGURE 30-1 Duodenal atresia (and stenosis) is depicted. In type I (A), either a membrane (B) or web (C) causes the intrinsic duodenal obstruction. There is no fibrous cord and the duodenum remains in continuity. Type II is characterized by complete obliteration of a segment of the duodenum with the proximal and distal portions attached via a fibrous cord. Type III is associated with complete separation of the dilated proximal duodenum from the collapsed distal duodenum.
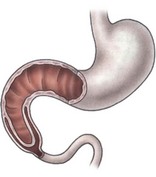
FIGURE 30-2 Illustration of the ‘windsock’ deformity, a variant of type I duodenal atresia. Note the actual position of the origin of the web in relation to the extent of proximal duodenal dilation and the distal collapsed duodenum.
More than 50% of affected patients with duodenal atresia have associated congenital anomalies.28 Approximately 30% are associated with trisomy 21, 30% with isolated cardiac defects, and 25% with other gastrointestinal (GI) anomalies.29,30 Approximately 45% of patients are premature, and about one-third exhibit growth retardation.6,7
Pathology
The obstruction can be classified as either preampullary or postampullary, with approximately 85% of obstructions located distal to the ampulla.30 With complete or almost complete obstruction, the stomach and proximal duodenum become significantly dilated. The pylorus is usually distended and hypertrophic. The bowel distal to the obstruction is collapsed, except in the case of a windsock deformity in which the distal bowel is dilated to a variable length depending on the length of the windsock (see Fig. 30-2). In most cases of duodenal obstruction, the gastrointestinal tract can be decompressed proximally. With complete obstruction of the duodenum, the incidence of polyhydramnios ranges from 32–81%.1,31–34 Growth retardation is also common, presumably from nutritional deprivation from the swallowed amniotic fluid.
Diagnosis
There are multiple benefits to the antenatal diagnosis of duodenal obstruction, including parental counseling. The diagnosis can often be suggested by prenatal ultrasound (US). Sonographic evaluation in fetuses of mothers with a history of polyhydramnios can detect two fluid-filled structures consistent with a double bubble in up to 44% of cases.35–37 Despite duodenal obstruction usually occurring by week 12, the reason for failure of early prenatal detection is not entirely clear. Most cases of duodenal atresia are detected between 7 and 8 months gestation.38 It is currently believed that immature gastric emptying in-utero may contribute to low gastric pressures, failing to dilate the proximal duodenum until later in gestation. While both circular and longitudinal muscle layers are present in the stomach by week 8 of gestation, pressure amplitudes at 25 weeks are only 60% of term gastric pressures.39,40
The presentation of the neonate with duodenal obstruction varies depending on whether the obstruction is complete or incomplete, and the location of the ampulla of Vater in relation to the obstruction. The classic presentation is that of bilious emesis within the first hours of life in an otherwise stable neonate. In about 10% of cases, however, the atresia is pre-ampullary and the emesis is nonbilious.15 Abdominal distention may or may not be present. In neonates with duodenal atresia, the abdomen is scaphoid. Aspiration via a nasogastric (NG) tube of more than 20 mL of gastric contents in a newborn suggests intestinal obstruction, as normal aspirate is less than 5 mL.41 For patients with stenosis, the diagnosis is often delayed until the neonate has started on enteral feeds and feeding intolerance develops with emesis and gastric distention.
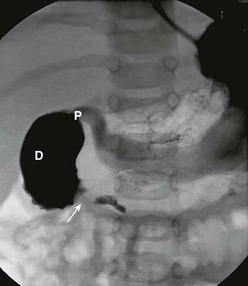
In antenatally suspected cases of duodenal obstruction, as well as in neonates with a clinical presentation consistent with a proximal bowel obstruction, an upright abdominal radiograph is usually sufficient to confirm the diagnosis of duodenal atresia. The diagnostic radiographic presentation of duodenal atresia is that of a double bubble sign with no distal bowel gas (Fig. 30-3). The proximal left-sided bubble represents the air and fluid-filled stomach while the dilated proximal duodenum represents the second bubble to the right of midline.42 In almost all cases of duodenal atresia, the distal bowel is gasless. However, the presence of distal gas does not necessarily exclude the diagnosis of atresia as there are reports of bifed common bile ducts with insertion of one of the ducts proximal and the other distal to the atretic segment which allows the air to bypass the atresia.43 In neonates whose stomach has been decompressed by either NG aspiration or vomiting, 40–60 mL of instilled air into the stomach will reproduce the double bubble.27 Rarely, the biliary tree is air filled, and a variety of pancreatic and biliary anomalies have been demonstrated (Fig. 30-4).43 At our institution, neonates who present with bilious emesis and a decompressed stomach on plain abdominal films receive a limited upper GI contrast study to exclude malrotation and volvulus. With duodenal stenosis, a double bubble sign is often not present and the diagnosis is usually made with a contrast study (Fig. 30-5).
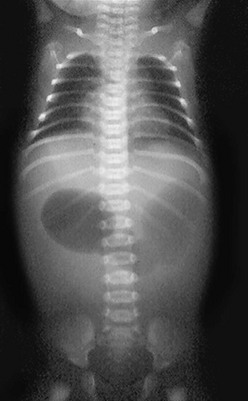
FIGURE 30-3 Classic ‘double bubble’ sign. This abdominal radiograph in a newborn shows a markedly distended stomach and duodenal bulb without evidence of distal intestinal air.
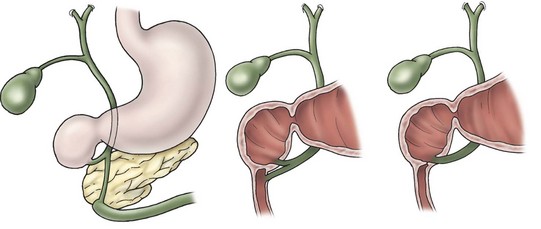
FIGURE 30-4 This schematic depicts several of the variations in biliary ductal anatomy seen in babies with duodenal atresia.
Management
After the diagnosis is made, appropriate resuscitation is required with correction of fluid balance and electrolyte abnormalities, in addition to gastric decompression. At our institution, all neonates diagnosed with duodenal obstruction receive a complete metabolic profile, complete blood count, coagulation studies, an abdominal and spinal ultrasound, and two-dimensional echocardiography prior to any operation. An emergency operation is only performed in cases where malrotation with concurrent volvulus cannot be excluded.
Prior to the mid-1970s, duodenojejunostomy was the preferred technique for correcting duodenal atresia or stenosis.7,44,45 Since then, the various techniques utilized include side-to-side duodenoduodenostomy, diamond-shaped duodenoduodenostomy, partial web resection with Heineke–Mickulicz-type duodenoplasty, and tapering duodenoplasty.44–46 The long side-to-side duodenoduodenostomy, although effective, is associated with a high incidence of anastomotic dysfunction and prolonged obstruction.30 Blind-loop syndrome appears to be more common in patients treated with duodenojejunostomy.47 Gastrojejunostomy should not be performed as it is associated with a high incidence of marginal ulceration and bleeding.27
Currently, the preferred technique is either laparoscopic or open duodenoduodenostomy.9–11,30 Originally, a side-to-side anastomosis was performed. A proximal transverse to distal longitudinal (diamond-shaped) anastomosis is now preferred.7,44,45,48–50 For the open approach, either a right upper quadrant supraumbilical transverse incision or an umbilical crease incision is utilized.49 After mobilizing the ascending and transverse colon to the left, the duodenal obstruction is readily exposed. Malrotation should be evaluated at this point as it can occur in association with congenital duodenal obstruction in up to 30% of patients.1 A sufficient length of duodenum distal to the atresia is mobilized to allow for a tension-free anastomosis. A transverse duodenotomy is made in the anterior wall of the distal portion of the dilated proximal duodenum and a similar length duodenotomy is made in a vertical orientation on the antimesenteric border of the distal duodenum. The anastomosis is then fashioned by approximating the end of each incision to the appropriate mid-portion of the other incision (Fig. 30-6). Tapering duodenoplasty is generally not necessary as the proximal duodenal dilation usually resolves after relief of the obstruction. Muscular continuity of the duodenal wall suggests a windsock deformity or diaphragm. This finding should precipitate extra vigilance in the operative correction because the dilated and collapsed bowel are both distal to the windsock, and have been anastomosed in error.26,51
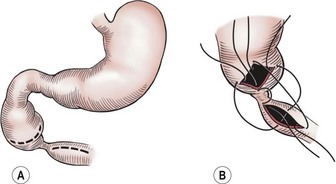
FIGURE 30-6 The technique of duodenoduodenostomy. A diamond-shaped anastomosis is created via the proximal transversely oriented and distal vertically oriented duodenotomies.
The laparoscopic approach was first described by Rothenberg in 2002.9 The standard laparoscopic approach begins with the patient supine and the abdomen is insufflated through the umbilicus. Two other instruments are inserted, one in the baby’s right lower quadrant and one in the right mid-epigastric region, respectively. A liver retractor can be placed in the right or left upper quadrant if necessary. Alternatively, the liver can be elevated by placing a transabdominal wall suture around the falciform ligament and tying it outside the abdomen (Fig. 30-7). The duodenum is mobilized and the location of obstruction is identified. Using the same principles that have been described for the open approach, a standard diamond-shaped anastomosis is created. Although some surgeons will perform the laparoscopic anastomosis with interrupted sutures, this can be technically demanding because of the significant number of sutures required and the thin nature of the distal duodenum. We reported our results using Nitinol U-clips (Medtronic, Minneapolis, MN) to create the duodenoduodenostomy with no leaks and more rapid initiation of feeds when compared to the traditional open approach (Fig. 30-8).10,11 Although the U-clips are no longer commercially available, this study highlighted the advantages of early feeding. The historical approach to enteral feeding following duodenal atresia repair involved a period of waiting for the gastric output to become less bilious and the volume of gastric drainage to decrease, indicating return of intestinal function. This report showed that the time spent waiting for the gastric output to decrease is likely not necessary as all of the patients undergong the laparoscopic duodenoplasty had initiation of feeds without adverse events after an upper gastrointestinal contrast study on day 5 revealed no leak. When compared to infants undergoing an open operation with the historical postoperative management mentioned previously, there was a marked reduction in hospitalization for the laparoscopically corrected infants, primarily due to the early feeding.

FIGURE 30-7 Two approaches to placement of the instruments for a laparoscopic duodenal atresia repair. (A) The two right-sided instruments are the primary working sites for the surgeon. The liver retractor (arrow) has been placed in the left midepigastric region. The falciform ligament has been elevated by a suture placed under it and tied over the red rubber catheter, which is used as a bolster. The suture (dotted arrow) exteriorized in the infant’s left upper abdomen was placed in the dilated proximal duodenum so that it could be easily manipulated. (B) This is a similar configuration except the instrument elevating the liver (arrow) is placed in the infant’s right upper abdomen rather than the left upper abdomen. The suture that was placed through the proximal dilated duodenum in A was not needed in this particular case.
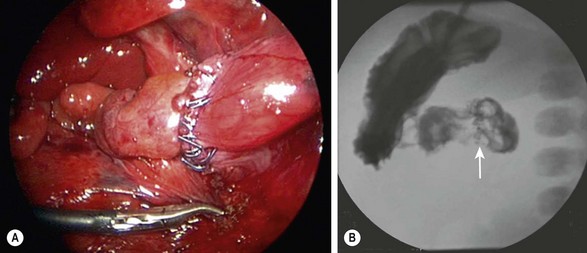
FIGURE 30-8 (A) Laparoscopic view of a completed duodenoduodenostomy using the Nitinol U-clips. (B) A postoperative contrast study at five days showed no evidence of obstruction or leak at the anastomosis. The U-clips (arrow) can be seen marking the anastomosis.
Historically, during repair of duodenal atresia, it has been emphasized that inspecting the entire small bowel to identify a second atresia is important. Given that duodenal atresia and jejunoileal atresia do not share common embryologic etiologies, a multi-institutional review of duodenal atresia patients was undertaken to quantify the incidence of jejunoileal atresia in this population.52 In the largest series to date, the rate of concomitant jejunoileal atresia in patients with duodenal atresia was less than 1%. With the low incidence of a concomitant distal atresia, extensive inspection of the entire bowel does not appear necessary.
Early postoperative mortality for duodenal atresia repair has been reported to be as low as 3–5% with the majority of deaths occurring secondary to complications related to associated congenital abnormalities.53,54 Long-term survival approaches 90%.7,53–55 Long-term complications have been noted following repair and include delayed gastric emptying, severe gastroesophageal reflux, bleeding peptic ulcer, megaduodenum, duodenogastric reflux, gastritis, blind-loop syndrome and intestinal obstruction related to adhesions.30
Jejunoileal Atresia and Stenosis
Etiology
Jejunoileal atresia occurs in approximately 1 in 5,000 live births. It occurs equally in males and females, and about one in three infants is premature.56 Although the majority of cases are thought to occur sporadically, familial cases of intestinal atresias have been described.57 It is generally accepted that jejunoileal atresia occurs as a result of an intrauterine ischemic insult to the midgut, affecting single or multiple segments of the already developed intestine.13,58–61 Intrauterine vascular disruption can lead to ischemic necrosis of the bowel with subsequent resorption of the affected segment or segments (Fig. 30-9).

FIGURE 30-9 The proposed mechanism of vascular compromise and subsequent development of jejunoileal atresias is depicted.
The hypothesis that most cases of jejunoileal atresia occur secondary to vascular disruption during fetal life is derived from experimental as well as clinical evidence. Isolated mesenteric vascular insults and interference with the segmental blood supply to the small intestine were created in fetal dogs, and resulted in different degrees and patterns of intraluminal obstruction, reproducing the spectrum of stenosis and atresia found in humans.62–64 Moreover, the presence of bile, lanugo hair, and squamous epithelial cells from swallowed amniotic fluid distal to an atresia suggests that the atresia occurs subsequent to some event, but that at some time in gestation the intestinal lumen was patent, thus allowing passage of these contents. Additionally, atresias seen in association with other intrauterine vascular insults such as fetal intussusception, midgut volvulus, thromboembolic occlusions, transmesenteric internal hernias, and incarceration or snaring of bowel in an omphalocele or gastroschisis have contributed to wide acceptance of this hypothesis.58,64–68
The presence of associated extra-abdominal organ abnormalities in jejunoileal atresia is low (<10%) due to its occurrence later in fetal life and the localized nature of the vascular insult.69 Rarely, jejunoileal atresia has been found in patients with Hirschsprung disease, cystic fibrosis, malrotation, Down syndrome, anorectal and vertebral anomalies, neural tube defects, congenital heart disease, and other GI atresias.56,69,70 Methylene blue, previously used for amniocentesis in twin pregnancies, has been implicated in causing small bowel atresia.71
Although jejunoileal atresias are usually not hereditary, there is a well-documented autosomal recessive pattern of inheritance of multiple atresias.72 In these cases, intestinal rotation was normal, mesenteric defects were never observed, and lanugo hairs and squamous cells were not identified distal to the most proximal atresia. All these findings suggest an early intrauterine event. Survival is poor in these infants, even with successful bowel resection.
No correlations have been found between jejunoileal atresia and parental or maternal disease. However, the use of maternal vasoconstrictive medications, as well as maternal cigarette smoking in the first trimester of pregnancy, has been shown to increase the risk of small bowel atresia.73 Chromosomal abnormalities are seen in less than 1% of the patients with jejunoileal atresia.74
Pathology
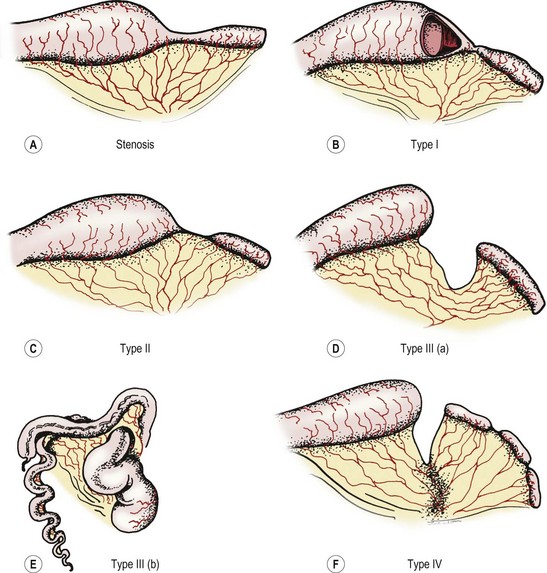
The Grosfeld classification system separates these defects into four groups, with an additional consideration for type III(b) (Fig. 30-10).4 This classification has significant prognostic and therapeutic value as it emphasizes the importance of associated loss of intestinal length, abnormal collateral intestinal blood supply, and concomitant atresia or stenosis.75 Regarding classification, the most proximal atresia determines whether the atresia is classified as jejunal or ileal atresia. Multiple atresias are found in up to 30% of patients.56,76
Stenosis
Stenosis is defined as a localized narrowing of the intestinal lumen without disruption in the intestinal wall or a defect in the mesentery (see Fig. 30-10A). At the stenotic site, a short, narrow, somewhat rigid segment of intestine with a small lumen is found. Often the muscularis is irregular and the submucosa is thickened. Stenosis may also take the form of a type I atresia with a fenestrated web. Patients with jejunoileal stenosis usually have a normal length of small intestine.
Type I Atresia
In type I jejunoileal atresia, the intestinal obstruction occurs secondary to a membrane or web formed by both mucosa and submucosa, while the muscularis and serosa remain intact (see Figs 30-10B and 30-11). On gross inspection, the bowel and its mesentery appear to be in continuity. However, the proximal bowel is dilated while the distal bowel is collapsed. With the increased intraluminal pressure in the proximal bowel, bulging of the web into the distal intestine can create a windsock effect. As with stenosis, there is no foreshortening of the bowel in type I atresias.
Type II Atresia
The clinical findings of a type II atresia are a dilated, blind-ending proximal bowel loop connected by a fibrous cord to the collapsed distal bowel with an intact mesentery (see Fig. 30-10C). Increased intraluminal pressure in the dilated and hypertrophied proximal bowel may lead to focal proximal small bowel ischemia. The distal collapsed bowel commences as a blind end, which sometimes assumes a bulbous appearance owing to the remains of an intussusception. Again, the total small bowel length is usually normal.
Type III(a) Atresia
In type III(a) atresia, the proximal bowel ends blindly with no fibrous connecting cord to the distal intestine. A V-shaped mesenteric defect of varying size is present between the two ends of intestine (see Figs 30-10D and 30-12). The dilated, blind-ending proximal bowel is often aperistaltic and frequently undergoes torsion or becomes overdistended, with subsequent necrosis and perforation occurring as a secondary event.77 In this scenario, the total length of the small bowel is variable (but usually less than normal), owing to intrauterine resorption of the affected bowel.