Disorders of Sexual Differentiation
INTRODUCTION
Normal sexual development requires a series of sequential and highly regulated events early in fetal development. This sexual differentiation process is regulated through complex interactions from both genetic and hormonal signals. Any disruption in this developmental pathway can lead to a disorder of sex differentiation (DSD).
Neonates born with ambiguous genitalia present a daunting challenge to all involved in health care delivery to the individual patient. An urgent evaluation by appropriate disciplines with discrete and delicate interaction with the family is mandatory. Older and potentially negative and confusing terminology has been replaced and should not be used. A new nomenclature for disorders of sex development, introduced at the 2005 Chicago Consensus Conference,1 is more precise, integrates better the progress in our understanding of the molecular basis of development, and is more sensitive to the concerns of the family and patients (Table 48-1).
Table 48-1 Proposed Revised Nomenclature
NORMAL SEXUAL DIFFERENTIATION AND DEVELOPMENT
Undifferentiated Stage of Sexual Differentiation
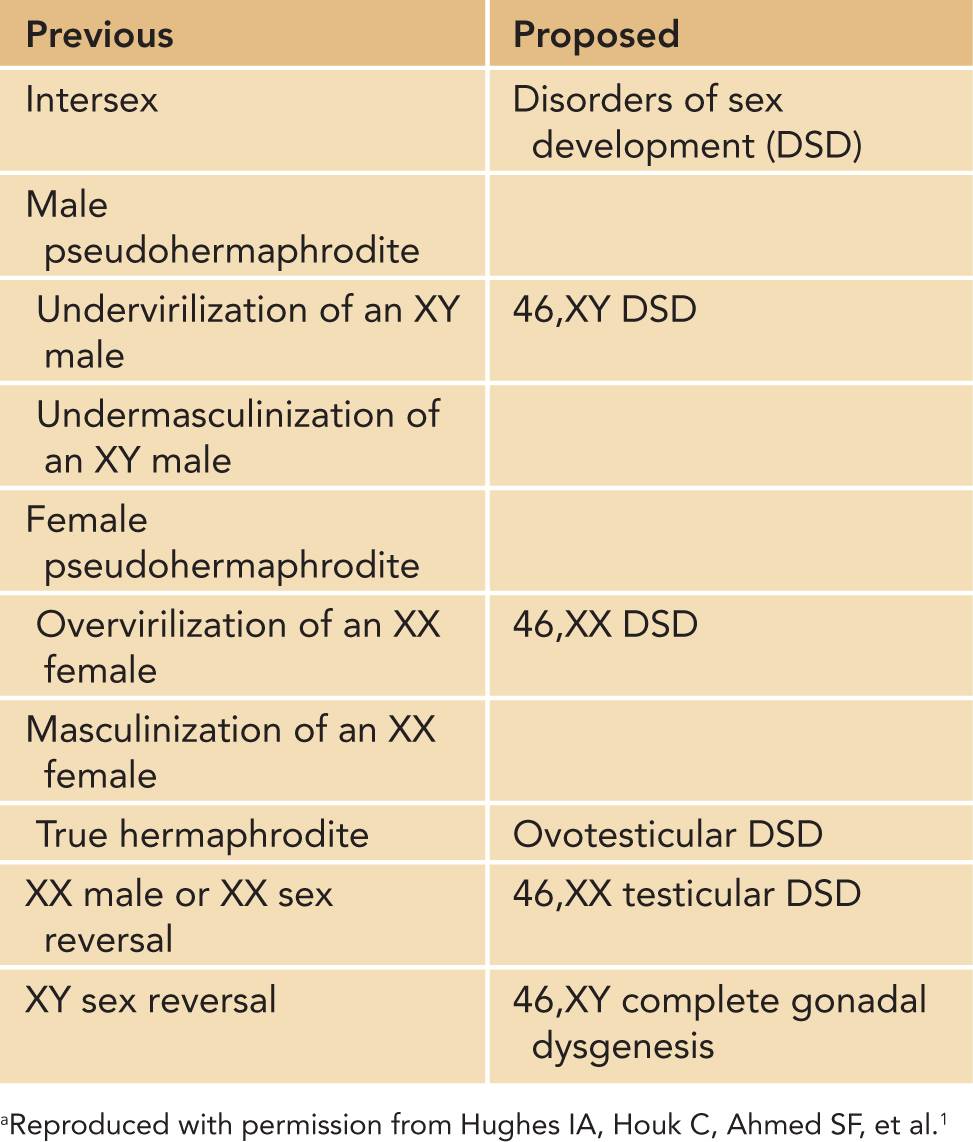
Normal fetal development of the gonads and the genitalia proceed through 3 sequential stages. The first stage involves the formation of the bipotential gonads from the adrenogonadal ridge at approximately 4 to 5 weeks’ gestation. During this undifferentiated stage, identical structures form in both the XY and XX embryo (Figure 48-1). These structures emanate from the mesonephros and coelomic epithelium. They are the wolffian (precursors to the formation of the seminal vesicles, epididymis, and vas deferens) and müllerian ducts (precursors to the fallopian tubes, uterus, and vagina).
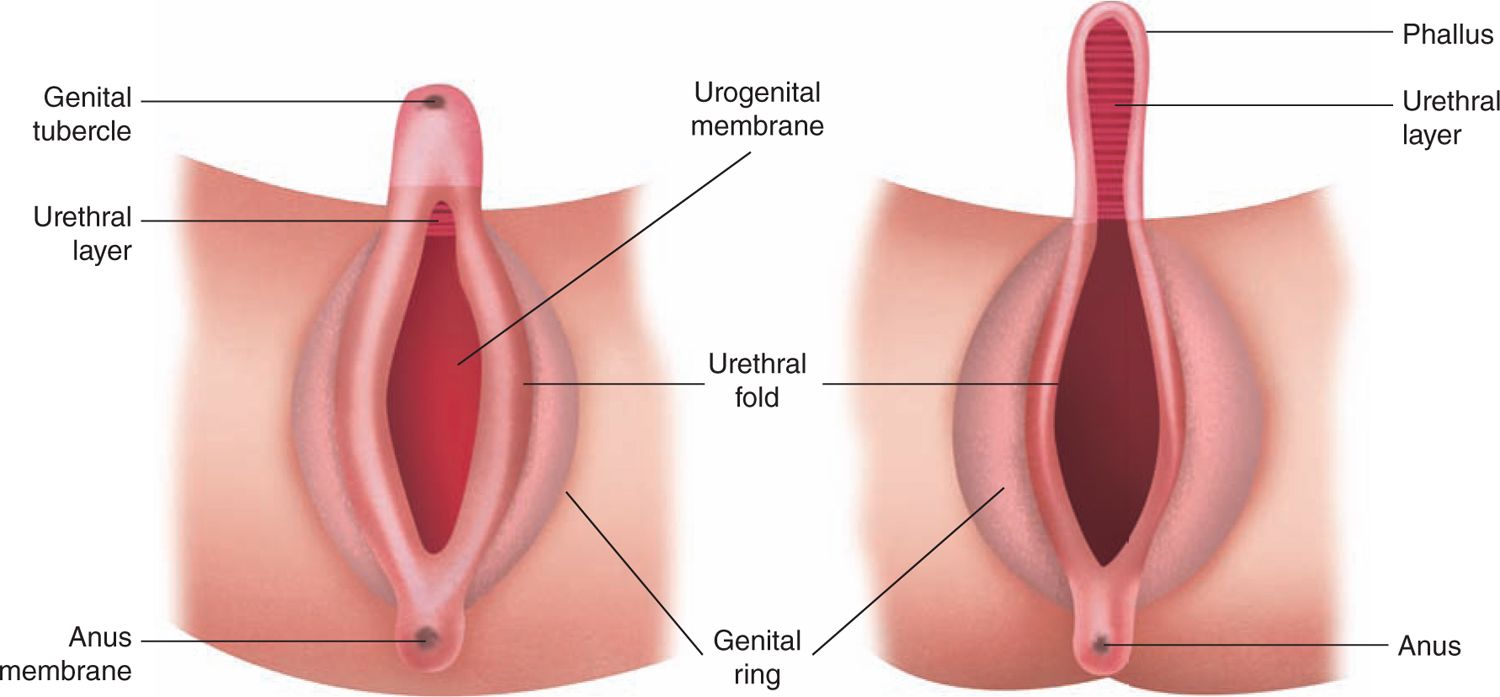
FIGURE 48-1 Diagram of the external genitalia in the undifferentiated period. (Reproduced with permission from Diamond.29)
During this phase, the cloaca, which is the terminal portion of the hindgut limited by the cloacal membrane, is partitioned into an anterior urogenital sinus and a posterior anorectal canal. Subsequently, the urogenital sinus becomes the bladder in both sexes and is the precursor for the prostate and proximal urethra in males and the entire urethra and vagina in females.
Gonadal Differentiation
Testis Formation
By the seventh week of gestation, the undifferentiated gonads in a male embryo develop into a testis under the influence of the SRY (sex determining region of the Y chromosome) gene located on the short arm of the Y chromosome. The SRY gene expression causes the primitive sex cords to differentiate into Sertoli cells, which when organized form the seminiferous cords. Primordial germ cells populate near the basement membrane of the cords, while Leydig cells appear in the interstitium between the cords around 8 weeks.
Ovary Formation
In the female embryo, the undifferentiated gonad does not contain the Y chromosome. Thus, there is no SRY gene production with no resultant differentiation of cells into Sertoli cells. The primitive sex cords degenerate, and secondary sex cords formed from the genital ridge join with primordial germ cells to form the ovarian follicles. At this time, the germ cells differentiate into oogonia and become oocytes following the first meiotic division. Many oocytes at this time are surrounded by a single layer of granulosa cells, becoming primary follicles at around 15 weeks’ gestation. Further meiosis is arrested under puberty when under hormonal influence meiosis resumes. The total population of germ cells reaches an apex of several million by 5 months’ gestation. However, by the time of birth, most cells have undergone apoptosis, leaving approximately 150,000 oocytes in each ovary at birth.
Other Genes Involved in Gonadal Differentiation
DAX1
The DAX1 gene has been implicated in several cases of XY sex reversal.2 Screening of XY females with a normal SRY gene detected the presence of a submicroscopic duplication of a region designated DDS (dosage-sensitive sex reversal). Within this region, the DAX1 gene was identified. Investigation using a transgenic, XY genotype mouse carrying extra copies of the DAX1 gene led to a phenotypically female mouse in the setting of low expression of the SRY gene.3
SOX9
The SOX9 gene was found to play a role in sex determination through investigation of patients with campomelic dysplasia (CD).4 In this rare skeletal disorder, a high preponderance of patients were found to have male-to-female sex reversal. Loss-of-function mutations of SOX9 were found in XY females with CD, pointing to a critical role SOX9 likely plays in sex determination.5 While the exact targets of SOX9 are unclear, it is structurally similar to SRY and has been proposed to be responsible for an SRY-negative, XX male patient.6
SF-1
Steroidogenic factor 1 (SF-1) is a transcription factor that plays a key role in the regulation of endocrine function and development of the adrenal glands and the gonads in both sexes. The results of SF-1 knockout mice models demonstrated a failure of adrenal and gonadal development with XY sex reversal with persistence of the müllerian ducts in males. Ovaries fail to develop in XX female mice lacking the SF-1 gene.7 Clinically, there has been a report of an XY female who was found to have adrenal failure at 2 weeks with a poorly differentiated testis, normal female genitalia, and the presence of a uterus. On evaluation, she was found to have a heterozygous mutation in the DNA-binding domain of SF-1, which was likely the cause of the clinical findings.8
WT1
The Wilms tumor 1 (WT1) gene was originally discovered on chromosome 11p13 during efforts to find the oncogene responsible for Wilms tumor.9 Following its discovery, further investigation of WT1 expression in humans and mice models demonstrated a significant relationship between WT1 protein secretion and sex determination. WT1 expression has been found to have a complex role with multiple sites of action, including modulating SF-1 expression, regulating SRY gene actions, and competing with DAX1.10–12
Mutations involving WT1 expression are linked to urinary and genital abnormalities in 3 syndromes: WAGR (Wilms tumor, aniridia, genitourinary anomalies, and mental retardation) syndrome, Denys-Drash syndrome, and Frazier syndrome. Denys-Drash syndrome is a rare genetic disorder associated with gonadal and genital abnormalities, Wilms tumor, and renal failure due to mesangial sclerosis. Frasier syndrome is characterized by male-to-female sex reversal, focal segmental glomerular sclerosis, and Wilms tumor.
Differentiation of the Genital Ducts and External Genitalia
Male Genital Duct Development
By the eighth week of gestation, the developing Sertoli cells secrete müllerian-inhibiting substance (MIS), which causes the müllerian ducts to regress rapidly by the 10th week of gestation. Between the 8th and 12th weeks, Leydig cells secrete testosterone, which stimulates the cranial aspect of the wolffian duct to transform into the epididymis and vas deferens. The caudal portion of the wolffian duct develops into the ejaculatory ducts and seminal vesicles (Figure 48-2).
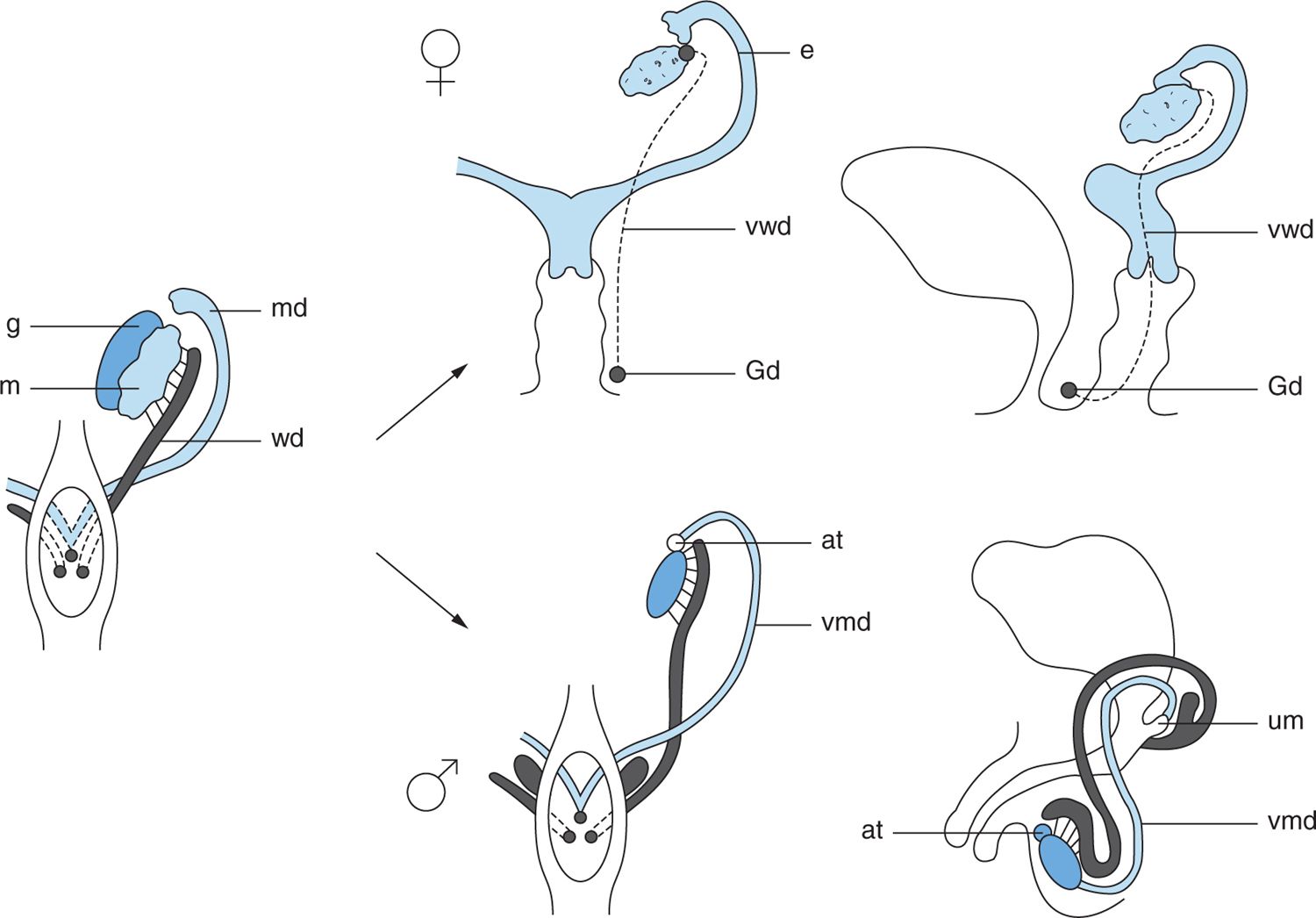
FIGURE 48-2 Fate of the wolffian and müllerian ducts. In the male, the wolffian ducts form the epididymis, vas, and seminal vesicle. In the female, the müllerian ducts form the fallopian tubes, uterus, and upper third of the vagina. at, appendix testis; e, epoöphoron; G, gonad; Gd, Gartner duct; m, mesonephros; md, müllerian duct; um, utriculus masculinus; vmd, vestigial müllerian duct; vwd, vestigial wolffian duct; wd, wolffian duct. (Reproduced with permission from Kelalis P, King LR, Belman AB, et al: The Kelalis-King-Belman textbook of clinical pediatric urology. 5th ed. Abingdon, UK: Informa Healthcare; 2007.)
Female Genital Duct Development
In the female fetus, müllerian duct formation proceeds in the absence of MIS secretion. The müllerian ducts give rise to the fallopian tubes and uterus by the 12th week of gestation. Historically, it was accepted that the upper two-thirds of the vagina originated from the müllerian ducts, with the lower third developing from the posterior urogenital sinus and fusing around 12 weeks’ gestation. Recent small-animal studies have called into question this theory, with genetic molecular work pointing to a complete müllerian vagina.13
Development of the External Genitalia
The early development of the external genitalia is similar in both sexes. Around the fifth week of gestation, a pair of swellings called the cloacal folds joins anterior to the cloacal membrane to form the genital tubercle. The urogenital and labioscrotal folds then appear shortly on either side of the genital tubercle.
Development of Male External Genitalia
In the male fetus, the undifferentiated stage of genital development ends around the ninth week with secretion of testosterone and local conversion to dihydrotestosterone causing an increase in distance between the genital tubercle and anal folds (Figure 48-3). The genital tubercle elongates to become the penis, with the urethral groove forming over the ventrum of the penis. Urethral folds, which are extensions of the urogenital folds, form the lateral margins of the urethral groove. The urethra is then created with the fusion of the urethral folds over the urethral groove. The labioscrotal folds then fuse in the midline to create the scrotum. The urogenital folds fuse over the newly created urethra to provide skin coverage for the penis. This process is completed by the 14th week. The phallus will continue to grow throughout the remainder of gestation. Testicular descent occurs through multiple mechanisms and is typically complete by the third trimester.
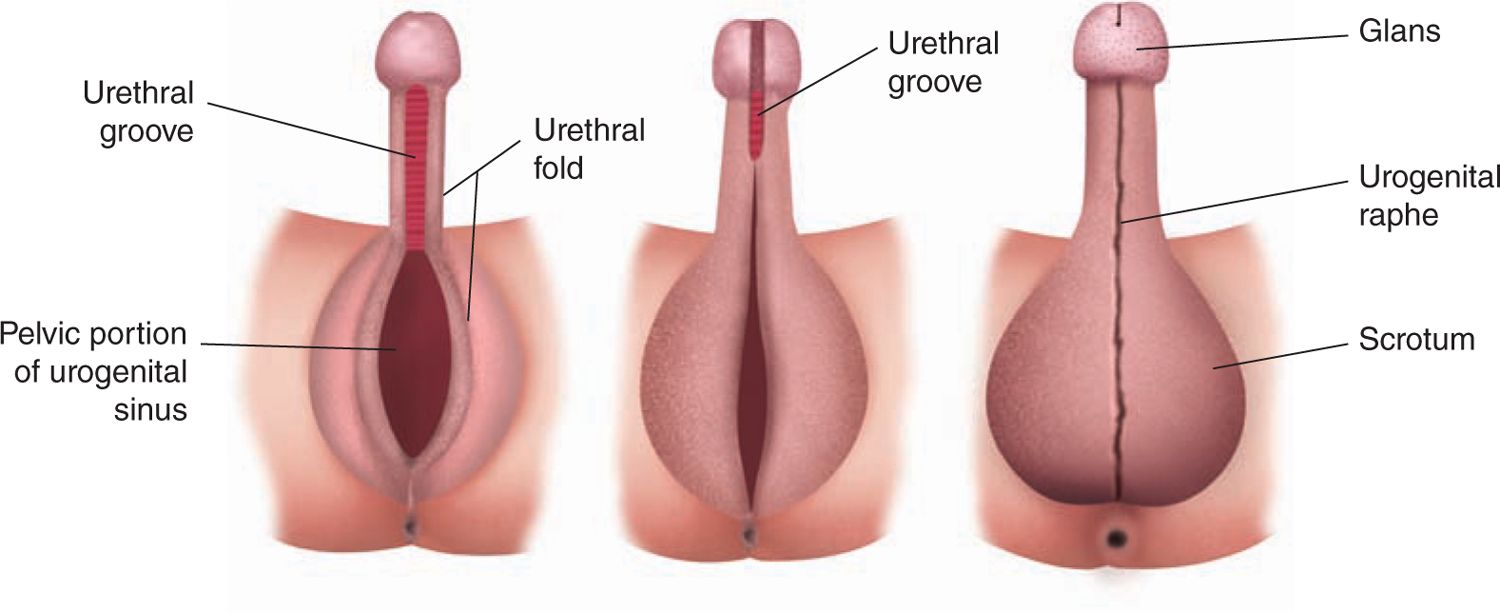
FIGURE 48-3 Differentiation of the male external genitalia. (Reproduced with permission from Diamond.29)
Development of Female External Genitalia
In the absence of testosterone, the genital tubercle bends inferiorly and forms the clitoris in the female fetus (Figure 48-4). The labioscrotal folds do not fuse in the midline and develop primarily in the caudal portions, forming the labia majora. The urethral folds do not fuse and become the labia minora. The anterior urethral meatus and posterior vaginal orifice form between the labia minora, with this process complete by 14 weeks.
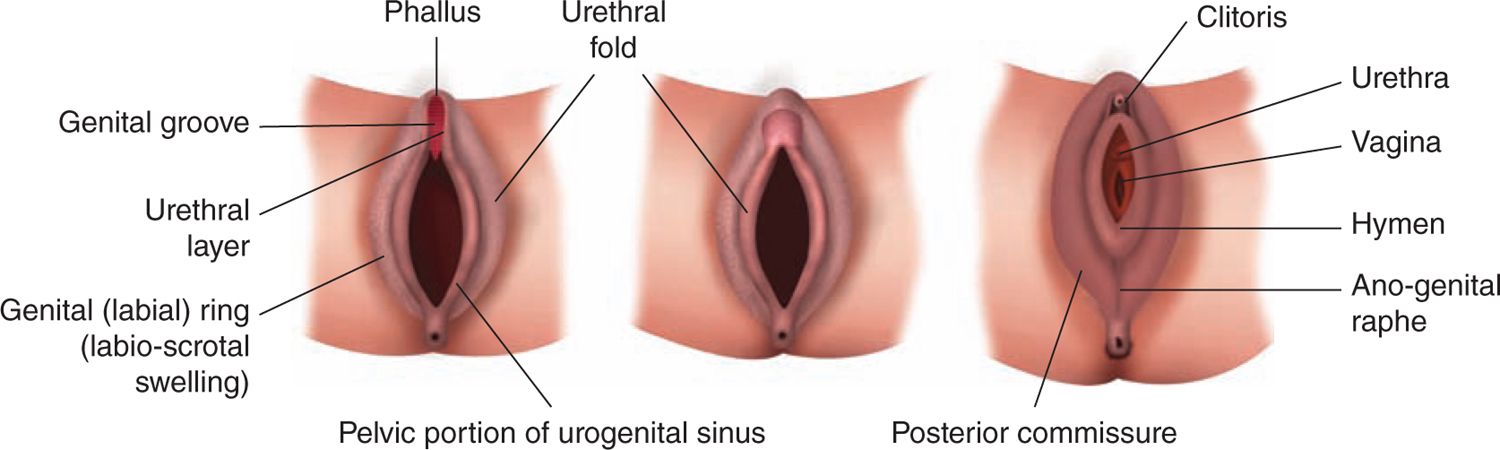
FIGURE 48-4 Differentiation of the female external genitalia. (Reproduced with permission from Diamond.29)
Psychosexual Differentiation
Our understanding of human psychosexual differentiation is limited and appears complex and multifactorial. The previously accepted view that humans are psychosexually neutral at birth14 has more recently been questioned by those who support the opposing view that a “neural bias” exists determined primarily by prenatal hormonal exposure.15 Studies in mammals demonstrated a significant role of both testosterone and estrogen exposure prenatally and sexual differentiation of the brain and behavior in utero.16,17 This relationship between hormone exposure in utero and psychosexual differentiation is less clear in humans. However, the accumulated evidence demonstrates that androgen exposure prenatally has more influence on gender role (aspects of behavior in which males and females appear to differ) than gender identity (identification of oneself as either male or female).18
CLINICAL EVALUATION OF INFANTS WITH AMBIGUOUS GENITALIA
Prenatal Findings
Prenatal suspicion of a DSD occurs most commonly when abnormalities of the genitalia are found on prenatal sonography or when there is a suspected genotype-phenotype mismatch with the karyotype and visualized genitalia or uterus.19 Characteristic findings are seen with ultrasound allowing for accurate prenatal sexual determination. Normal male genital findings on ultrasound include the penis, which is initially visualized as an upwardly projected phallic structure, in contrast to the clitoris, which will have a downward bend.19 Also, fetal penile length can then be measured in terms of a range throughout development.20 The scrotum can also be detected as a distinct structure by the second trimester21 and is sonographically distinct from the developing labia.22 In addition, a nomogram exists for uterine width and circumference measurements, which allows for assessment of normal female genital tract development from the second trimester until birth.23
In the rare situations for which genital abnormalities are suspected prenatally, it is important to follow the fetus longitudinally throughout the pregnancy. Fetal ultrasounds at 13–15 weeks are not good predictors of external genital development.19 The presence of a uterus after 19 weeks is a reliable predictor of internal reproductive organs. Karyotypic or external genital discrepancies based on the presence of a uterus should lead to cautious antenatal counseling, which can include the disciplines of neonatology, pediatric endocrinology, genetic counseling, pediatric urology, and psychology. Certainly, the assignment of sex for the child would be withheld until appropriate postnatal evaluation.
Postnatal Evaluation
Every newborn must have a careful and complete genital examination. The importance of the examination includes not only assignment of the appropriate gender, but also the accurate diagnosis of more common abnormalities, such as hypospadias, chordee, and undescended testes.
Optimal management of neonates with a suspected DSD as recommended by the Lawson Wilkins Pediatric Endocrine Society (LWPES) and the European Society for Paediatric Endocrinology (ESPE) includes the following: (1) Avoidance of gender assignment before expert evaluation is complete. (2) Evaluation and long-term management must be carried out at center with an experienced multidisciplinarian team. (3) All neonates should receive a gender assignment. (4) Open communication with patients and families is essential, and participation in decision making is encouraged. (5) Patient and family concerns should be respected and addressed in strict confidence.
Physical Examination
In the apparent male neonate, the palpability, location, and size of the testis (or ovotestis) should be noted. Ovaries do not descend. The length and diameter of the penis should be measured. Penile length is measured as the stretched length following the reduction of the prepubic fat from the pubic ramus to the tip of glans. Boys with a stretched penile length at term of less than 2 cm in an otherwise physically normal phallus have a micropenis. The location of the urethral meatus should be properly identified. The presence of ventral congenital curvature (chordee) should be documented. Neonates with severe chordee can make obtaining a true stretched penile length difficult. A flat, symmetric scrotum typically indicates bilateral undescended testes. A cleft or bifid scrotum usually occurs in conjunction with a severe penoscrotal or scrotal hypospadias and can look similar to labia.
In the apparent female neonate, the clitoris should be assessed for general size. Clitoromegaly can be dramatic, and in these patients, a stretched length and width should be measured. The urethral location and relationship to a separate vaginal orifice should be carefully assessed. In female fetuses exposed to high levels of testosterone in utero, a common distal stem off the vagina and urethra can occur (urogenital sinus). The os of the urogenital sinus can be located on the perineum or associated with the masculinized clitoris. The labia are normally unfused in females. Fusion, anterior placement of the labia with hyperpigmentation, and absence of labia minora are also signs of in utero androgen exposure. The uterus may be palpable on rectal examination as a cord-like structure anterior to the rectum.
A DSD evaluation should proceed in neonates with the physical findings of (1) overt genital ambiguity; (2) apparent female genitalia with an enlarged clitoris, posterior labial fusion, or an inguinal or labial mass; or (3) apparent male genitalia with bilateral undescended testes, micropenis, isolated perineal hypospadias, or hypospadias (Figure 48-5).
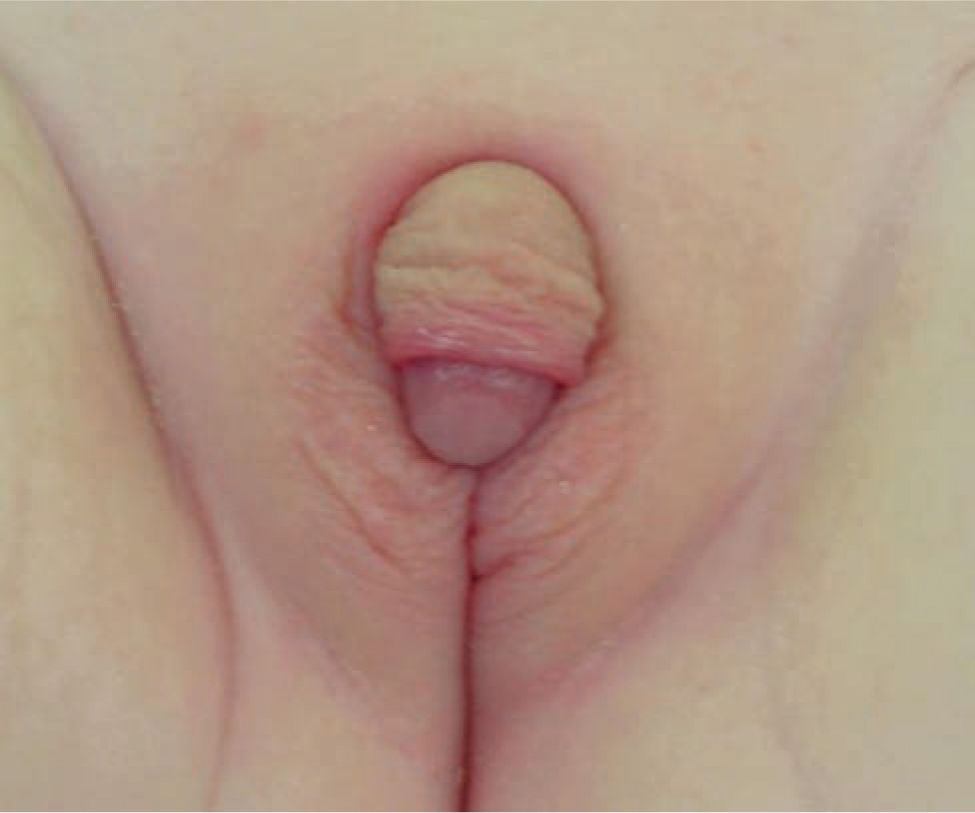
FIGURE 48-5 Virilized female genitalia.
Laboratory Evaluation
Immediate laboratory evaluation of a neonate with a suspected DSD consists of measurement of serum electrolytes, testosterone, and dihydrotestosterone levels and karyotype. Serum 17-hydroxyprogesterone should not be measured until day of life 3 or 4 because elevated levels of this corticosteroid precursor can be found in normal neonates during the first few days of life due to the stress of delivery.24 If 17-hydroxyprogesterone is elevated, 11-deoxycortisol and deoycorticosterone levels are required. LH (luteinizing hormone) levels or a hCG (human chorionic gonadotropin) stimulation test can be performed to determine if functioning testicular tissue is present in the neonate with bilateral nonpalpable testes.
Radiographic Evaluation
An abdominal and pelvic ultrasound is recommended as the first-line imaging study in the neonate with ambiguous genitalia. The presence of a uterus is accurately seen with pelvic ultrasound.25 Gonads may be seen, but the specificity and sensitivity of ultrasound is poor for nonpalpable testes and the presence of ovaries.26 The absence of visible gonads does not confirm their absence. In experienced hands, adrenal sonography is an accurate adjunct for the diagnosis of congenital adrenal hyperplasia (CAH).27 Studies, such as pelvic magnetic resonance imaging (MRI), flush genitogram, and retrograde urethrogram, are necessary in certain situations, typically dependent on physical examination findings.
CLASSIFICATION OF DISORDERS OF SEXUAL DIFFERENTIATION
Sex Chromosome Disorder of Sex Development
45,X/46,XY (Mixed Gondal Dysgenesis)
Mixed gonadal dysgenesis (MGD) is a group of disorders characterized by a unilateral testis, which is typically undescended; an intra-abdominal contralateral streak gonad, persistent müllerian duct structures; and varying degrees of virilization of the external genitalia.28 Infants typically present with ambiguous genitalia as this condition is the second-most-common cause of a DSD in the newborn period.29 The unilateral testis may be descended but usually is palpable in the groin or nonpalpable when located abdominally.
The uterus is typically rudimentary with bilateral fallopian tubes. In those patients with a well-differentiated testis, the ipsilateral fallopian is commonly absent. A urogenital sinus is present in about one-half of patients.
When the unilateral testis is palpable, it is typically more normal. However, the testicular cellular architecture is consistently abnormal in the adult patient, demonstrating few germ cells and sclerosis of the tubules.30 Wolffian structures are more commonly found on the side of the unilateral testis and usually include only the epididymis, with a well-differentiated vas deferens rarely present.
47,XXY (Klinefelter Syndrome and Variants)
Klinefelter syndrome is the most common abnormality of sexual differentiation, occurring in approximately 1 in every 600 live newborn males.31 The syndrome is classically characterized by gynecomastia, small testes, absent spermatogenesis, normal-to-moderately reduced Leydig cell function, and increased secretion of follicle-stimulating hormone (FSH).32 It is most commonly associated with the 47,XXY karyotype, but upward of 20% of patients will have higher-grade chromosome aneuploidies, 46XY/47XXY mosaicism, or structurally abnormal X chromosomes.33
The diagnosis is made prenatally in approximately 10% of patients because of genetic screening programs.31 Neonatal diagnosis is rare but can occur during an evaluation for micropenis, severe hypospadias, and undescended or small testes. Typically, the diagnosis is made following puberty when androgen deficiency becomes apparent. Young men with Klinefelter syndrome have sparse body and facial hair. Approximately half of the patients will develop gynecomastia. Adult men have small testes, which rarely exceed 2 cm in length and are atrophic on microscopic examination.
45,X (Turner Syndrome and Variants)
Turner syndrome is associated with an immature female phenotype with short stature, streak gonads, and various congenital anomalies. It is classically and most frequently associated with a missing X chromosome. Variant forms occur less commonly and include partial deletions of the second X chromosome. Various mosaicisms do occur and typically involve the X chromosome. In approximately 5% of patients, a 45,X/46XY mosaicism is found.
Neonates with Turner syndrome do not have ambiguous genitalia but have other somatic features that are noted both antenatally and perinatally, which can lead to the diagnosis (Table 48-2). A clinical guideline pathway exists for those children with a new diagnosis of Turner syndrome. The workup includes a cardiology evaluation, renal ultrasound, hearing testing, and referral to the appropriate support groups. Those patients with a diagnosis missed early in life are typically diagnosed later because of short stature, primary amenorrhea, or lack of secondary sexual development.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


