Disorders of Sexual Differentiation
Normal Gender and Sexual Differentiation
The most commonly accepted paradigm, described by Jost, involves a stepwise process to gender and sexual development.1 The primary determinant is the chromosomal gender, which is established at fertilization when the sperm provides an X or Y chromosome to the ovum’s X chromosome. Chromosomal gender determines gonadal gender, with XX resulting in ovarian development and XY resulting in testicular formation. Finally, the gonadal function determines the phenotypic gender. Although this paradigm is a helpful cascade to explain gender development, the simple Y = male, no Y = female equations are not always valid.
The testis determining factor (TDF) is located on the short arm of the Y chromosome near the centromere at the distal aspect of the Y-unique region.2 TDF is a 35 kb pair sequence on the 11.3 sub-band of the gender-determining region of the Y chromosome (SRY). Interestingly, SRY appears to be expressed by the somatic cells from the urogenital ridge and not from germ cells.
Many other genes play a role in gender development. Despite the presence of a functional SRY gene, the absence of the SOX9 gene results in a female phenotype in the majority of chromosomal males.3,4 The Wilms tumor gene (WT1) appears to play a key role not only in renal development, but also in testicular development. Early alteration of WT1 function results in testicular agenesis and later dysfunction results in aberrant testicular development (streak gonad or dysgerminomas). This tumor suppressor gene has been implicated in Denys–Drash syndrome involving testicular (mixed gonadal dysgenesis) and renal (Wilms tumor) abnormalities.5,6
Fushi–Tarzu factor-1 (FTZ-F1) exerts its effect on gonadal development through its regulation of steroidogenic factor-1 (SF-1). The SF1 gene is involved with steroid hormone production and the production of Müllerian-inhibiting substance (MIS) by the Sertoli cells of the testis that causes regression of the Müllerian ductal system. Although FTZ-F1 and SF-1 are also expressed in ovarian tissues, the timing and intensity of their effect are critical for normal gonadal development.5,7
Finally, the lack of an SRY gene alone does not impart normal female phenotypic and gonadal development. The DAX1 gene appears to be essential for the development of the ovary. The DAX1 gene product appears to compete with the SRY gene product for a steroidogenic regulatory protein (StAR). A dosage-sensitive element is also important. Normally, the single SRY gene has a greater impact than a single DAX1 gene and causes upregulation of StAR. However, in those chromosomal abnormalities in which more than one DAX1 gene is present, downregulation of StAR occurs, testicular development is inhibited, and ovarian development is promoted. As in the case of Turner syndrome, these primordial ovaries develop into streak gonads. Likely, other genes are also important for normal ovarian development.8–10
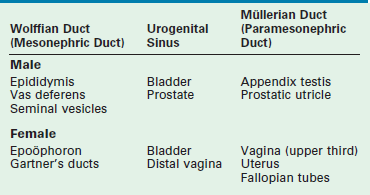
Development of the internal ductal structures is dependent on hormone secretion by the developing gonads (Table 62-1). In the absence of functioning testicular tissue, the female internal Müllerian duct structures develop. The presence of a functioning testis results in male internal Wolffian duct development. This differentiation is mediated by the production of testosterone from the testis. Testosterone promotes Wolffian duct development along with MIS, which results in regression of the Müllerian duct structures. This is a paracrine effect and therefore results in gonad specific ipsilateral ductal differentiation. This effect is likely dependent on high concentrations of androgen produced by the physically proximate gonad. Decreased levels of MIS by an abnormal testis or streak gonad result in ipsilateral Müllerian development. This occurs despite regression of the Müllerian ducts on the contralateral side with normal testicular MIS production. Conversely, systemic administration of androgen does not result in male ductal development in a female fetus.
Produced by Sertoli cells, MIS functions as a suppressor of Müllerian duct development and is a specific marker for functioning testicular tissue in infancy. In its absence, the Müllerian structures develop. The concentration and timing of MIS secretion appear to be critical. Normally, secretion occurs during week 7 of gestation. By week 9, the Müllerian ducts become insensitive to MIS.9
External genital development follows a similar path (Fig. 62-1). In the absence of the testosterone metabolite dihydrotestosterone (DHT), the external genitalia develop into the female phenotype. The male and female phenotypes are identical until week 7. In the male, testosterone production by the testicular Leydig cells surges at 7 weeks and remains elevated until week 14 of gestation. Testosterone is converted to DHT by 5α-reductase in the tissues of the genital skin and urogenital sinus. The testosterone-binding receptor has much higher affinity for DHT than testosterone and serves to amplify the effect of testosterone on the developing external genitalia. In the absence of 5α-reductase, the internal Wolffian ducts are preserved, but the external structures are feminized.

Aberrant Gender Development
Incidence
In the Americas and Western Europe, congenital adrenal hyperplasia (CAH) is the most common cause of neonatal ambiguous genitalia, accounting for approximately 70% of cases.11,12 The overall incidence is approximately 1 in 15,000 live births. The rate is much higher in stillborns and in certain regional populations (Yupic Eskimos and the people of La Réunion, France).13 In the United States, mixed gonadal dysgenesis is the next most common intersex disorder, with ovotesticular DSD (true hermaphroditism) being the third most common.
Classification
Until recently, the most commonly used classification system was the one proposed by Allen in 1976 and was based primarily on gonadal histology.14 This system categorized the most common DSD well, but did not accommodate less common syndromes easily. Also, the older ‘hermaphrodite’ terminology has become offensive to some. A newer classification released by the International Consensus Conference on Intersex has largely replaced Allen’s system. This newer system incorporates an evolving understanding of the molecular basis of these disorders and replaces more offensive gender-based labels. The new terminology is used primarily in this text.15
46,XX DSD (Female Pseudohermaphrodite)
The majority of neonates with external genital ambiguity fall into this category. All patients have a 46,XX genotype and exclusively ovarian tissue in nonpalpable gonads. Simplistically, the cause of the gender ambiguity is an excess of androgen. More than 95% are due to CAH, with the remainder due to maternal androgen exposure. These patients have a normal female Müllerian ductal system with an upper vagina, uterus, and fallopian tubes (see Table 62-1). They also have normal regression of the Wolffian ducts. The level of virilization is largely dependent on the timing and magnitude of androgen exposure to the external genitalia. The phenotype can range from mild clitoromegaly to a normal male appearance.
Virilization in CAH is due to the inability of the adrenal gland to form cortisol. The precursors above the enzymatic defect are shunted into the mineralocorticoid or sex-steroid pathways. Also, the end products generally have some, albeit weak, glucocorticoid function. The lack of cortisol for negative feedback inhibition of adrenocorticotropic hormone (ACTH) production by the pituitary leaves this pathway unchecked. Excess androgen is produced and is responsible for the virilization. The corticosteroid synthetic and alternative pathways are shown in Figure 62-2. The most common form of CAH is 21-hydroxylase deficiency (21-OHD), which accounts for more than 90% of CAH.16 21-OHD has been mapped to the short arm of chromosome 6. The variable location of the adrenal defect and relative function of the gene results in salt-wasting and nonsalt-wasting forms.17–19 Type 1 results in virilization but no salt wasting. The gene defect affects only the fasciculata zone of the adrenal, resulting in blocking cortisol production. However, the gene is normally expressed in the glomerulosa zone with preservation of mineralocorticoid production. In type 2, also called the classic type, the gene abnormality affects both adrenal zones. Salt wasting results in dehydration or vascular collapse, and hyperkalemia develops because of the block in mineralocorticoid production.
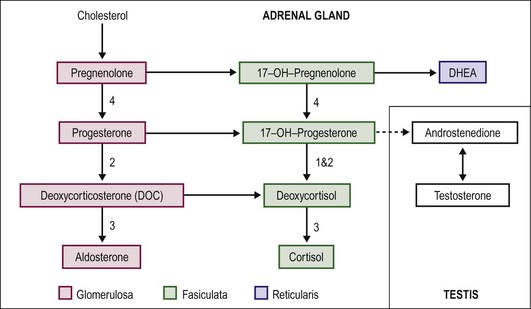
FIGURE 62-2 Pathways of steroid biosynthesis. The numbers correspond to CAH type and the location of the enzyme defect (see the text).
Virilization of the female fetus can be caused by exogenous androgen exposure from the mother. This occurs primarily with the use of progesterone, commonly used as an adjunct to assist with fertility and in-vitro fertilization. Endogenous androgen exposure due to virilizing maternal ovarian tumors also has been reported, but these tumors are usually virilizing to the mother with the fetus being unaffected.20
Treatment
As all forms of CAH are inherited in an autosomal recessive manner, genetic counseling is recommended. Families with a history of CAH should consider maternal treatment with dexamethasone before week 10 of gestation to eliminate or improve the level of fetal virilization.21 Postnatally, cortisol replacement with hydrocortisone is the mainstay of therapy, with the addition of fluorohydrocortisone if salt wasting is present. Supportive management of fluid and electrolyte abnormalities is best provided in a neonatal intensive care unit.
With regard to gender identity, the vast majority of patients with CAH identify as female and gender assignment is generally female given the 46,XX karyotype. The ovaries are normal and have fertility potential.22 Surgical reconstruction requires a feminizing genitoplasty, and involves clitoroplasty, monsplasty, and vaginoplasty.
46,XY DSD (Male Pseudohermaphrodite)
Defects in 17,20-desmolase and 17β-hydroxysteroid oxidoreductase act at the testicular level to convert androstenedione to testosterone. Because the adrenal is unaffected, CAH does not occur. The phenotype can be quite variable, but those with complete feminization can escape detection at birth and be reared as female. Progressive virilization is related to excess gonadotropin production at puberty, which may partially compensate for the lack of testosterone synthesis. Phallic growth and the development of male secondary sex characteristics create a conundrum with regard to gender reassignment when the diagnosis is made later in life.23
The extreme is normal female external genitalia resulting from complete androgen insensitivity syndrome (CAIS, also termed testicular feminization). The incidence of this syndrome is approximately 1 in 40,000. It usually results from a point mutation in the androgen receptor gene, located on the X chromosome.24,25
Partial androgen insensitivity is associated with a large spectrum of phenotypic variation (e.g., Gilbert–Dreyfus, Lub, and Reifenstein syndromes). It can be a sporadic or inherited condition, and gender assignment and treatment are individualized.26
Diagnosis
Metabolically, the diagnosis of CAH is made similarly to the 46,XX DSD patient, noting excess steroid levels above the enzymatic block and elevated levels of ACTH. The physical examination confirms absence of a cervix on rectal examination, and bronzing of the skin may be present. Palpation of a cryptorchid or descended testis is possible. The genitogram and ultrasound mirror these findings, but a prominent utricle may be present that lacks a cervical impression at its apex. In CAIS, testosterone levels are elevated postpubertally, but the diagnosis in the prepubertal child may require human chorionic gonadotropin (hCG) stimulation and genital skin fibroblast androgen receptor studies. Receptor assays can delineate a quantitative versus qualitative receptor defect. LH levels are elevated, related to the loss of testosterone feedback inhibition, which requires normal receptor hormone interaction. 5α-reductase deficiency is confirmed by an elevated testosterone-to-DHT ratio and an abnormal 5α-reductase type 2 gene.27
Treatment
In CAIS, the gender assignment is always female. CAIS patients who are assigned as female in infancy later identify themselves as female.28 Because the androgen receptor defect is ubiquitous, virilization of the brain does not occur. Orchiectomy is required, given the risk of malignant degeneration, but is often deferred until after puberty.29 The testis synthesizes estradiol, facilitating feminine development at puberty. Orchiectomy before puberty necessitates hormone replacement for normal pubertal development.
Gender assignment in partial androgen insensitivity syndrome (PAIS) is largely based on the response of the external genitalia to exogenous testosterone. A significant virilization response argues for the male gender. If there is no response, the female gender is favored. This subgroup is the most variable and has the least consensus with regard to gender assignment. There are reports of gender reassignment at puberty.30,31 Dissatisfaction with the gender of rearing occurs in approximately 25% of PAIS patients, whether raised male or female.32 In 5α-reductase deficiency syndrome, the brain is normally virilized and these individuals identify with the male gender. Thus, male gender assignment is recommended.33
Müllerian-Inhibitory Substance Deficiency (Hernia Uterine Inguinale)
MIS is produced by the Sertoli cells in the testis and causes regression of the Müllerian ductal structures. In this rare syndrome of abnormal MIS production or MIS-receptor abnormality, Wolffian ductal development is unimpaired, but the Müllerian ducts also persist. Because the infant has a normal male phenotype, this syndrome is rarely encountered in the neonatal period. The most common presentation to the surgeon is that of finding a fallopian tube adjacent to an undescended testis in the hernia sac at the time of orchiopexy.34
If this scenario is encountered, a biopsy of the gonad should be performed, the hernia should be repaired, and all structures left intact until completion of a full evaluation with karyotype and MIS levels. Apparent males can also present with bilateral nonpalpable testes, and Müllerian structures are found at laparoscopy (Fig. 62-3
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


