Disorders of Sexual Development (DSD)
Melvin M. Grumbach
Abnormalities of sex differentiation or disorders of sexual development (DSD) are divided into two broad categories: (1) errors in (primary) sex determination, such as sex chromosome anomalies and gene mutations that interrupt or disrupt normal gonadogenesis, which lead to abnormalities of gonadogenesis, and (2) errors in sex differentiation that cause abnormal development of the somatic sex structures—the genital ducts, urogenital sinus, and external genitalia. Intrinsic or extrinsic factors that adversely affect any of the stages of these mechanisms can cause anomalies of sexual structure. Table 539-1 presents the consensus Classification of Disorders of Sex Development for clinical use. Older terminology used for classification (shown in parentheses in Table 539-1) have been abandoned and replaced by newer designations.1,2 The management approach to these disorders, including discussion of gender determination, is discussed in the final section of this chapter.
46, XX DISORDERS OF SEXUAL DEVELOPMENT (FEMALE PSEUDOHERMAPHRODISM)
Persons with 46,XX disorders of sexual development have a 46,XX karyotype, ovaries, female ducts, and variable degrees of masculine differentiation of the urogenital sinus and external genitalia. The disorders are subdivided into those that are androgen induced and nonandrogen induced as shown in Table 539-1.
 CONGENITAL VIRILIZING ADRENAL HYPERPLASIA
CONGENITAL VIRILIZING ADRENAL HYPERPLASIA
This disorder is caused by an inborn error of adrenocortical hormone biosynthesis that results in relative deficiency of cortisol production, increased secretion of corticotropin (ACTH), and relative excess of androgenic hormones and other steroids. Diagnosis and management of these disorders is discussed in greater detail in Chapter 531. Congenital virilizing adrenal hyperplasia accounts for approximately one half of all cases of ambiguous external genitalia with the defect in 21-hydroxylation being by far the most common. The mode of inheritance is autosomal recessive.
Table 539-1. Classification of Disorders of Sex Development
Disorders of Gonadal Differentiation (Sex Chromosome DSD) |
Seminiferous tubule dysgenesis (Klinefelter syndrome) |
Syndrome of gonadal dysgenesis and its variants (Turner syndrome) |
Complete and incomplete forms of XX and XY gonadal dysgenesis including XX seminiferous tubule dysgenesis (XX phenotypic males) (eg, XX testicular DSD [SRY+; RSPO1; SOX9 duplication]) (see III-D for XY testicular dysgenesis) |
Individuals with both testicular and ovarian tissue: ovotesticular DSD (true hermaphroditism*) |
46,XX DSD (Female Pseudohermaphroditism*) |
Androgen induced |
Fetal source |
Congenital virilizing adrenal hyperplasia (defective 21-hydroxylation, 11β-hydroxylation, or 3β-hydroxysteroid dehydrogenase-2) |
Glucocorticoid receptor mutation |
Fetoplacental source |
P450 aromatase deficiency |
P450 oxidoreductase deficiency |
Maternal source |
Iatrogenic |
Testosterone and related steroids |
Certain synthetic oral progestagens |
Virilizing ovarian or adrenal tumor |
Virilizing luteoma of pregnancy |
Congenital virilizing adrenal hyperplasia in mother** |
Nonandrogen induced |
Disturbances in differentiation of urogenital structures associated with malformations of intestine and lower urinary tract (non-androgen-induced XX, DSD) |
46,XY DSD (Male Pseudohermaphroditism*) |
Testicular unresponsiveness to hCG and LH |
Leydig cell agenesis or hypoplasia |
Inborn errors of testosterone biosynthesis |
Enzyme deficits affecting synthesis of both corticosteroids and testosterone (variants of congenital adrenal hyperplasia) |
StAR deficiency (congenital lipoid adrenal hyperplasia): side chain (P450scc) cleavage deficiency |
3β-Hydroxysteroid dehydrogenase-2 deficiency |
P450c17 (17α-hydroxylase) deficiency |
P450 oxidoreductase deficiency |
7-dehydrocholesterol reductase deficiency (Smith-Lemli-Opitz syndrome) |
Enzyme defects primarily affecting testosterone biosynthesis by the testes |
P450c17 (17,20 lyase) deficiency |
17β-Hydroxysteroid dehydrogenase-3 deficiency |
Defects in androgen-dependent target tissues |
End-organ resistance to androgenic hormones (androgen receptor and postreceptor defects) |
Syndrome of complete androgen resistance and its variants (testicular feminization and its variant forms) |
Syndrome of partial androgen resistance and its variants (Reifenstein syndrome) |
Androgen resistance in infertile men |
Androgen resistance in fertile men |
Defects in testosterone metabolism by peripheral tissues 46,XY DSD |
5α-Reductase-2 deficiency (pseudovaginal perineoscrotal hypospadias) |
Dysgenetic 46,XY DSD |
XY gonadal dysgenesis (incomplete) |
XO/XY mosaicism, SRY mutation structurally abnormal Y chromosome, Xp+, 9p−, 10q− |
Denys-Drash Frasier syndrome (WT-1 mutation) |
WAGR syndrome (WT-1 deletion) |
Campomelic dysplasia (SOX9 mutation) |
SF1 mutation |
WNT-4 duplication |
DHH (mutation) |
ATRX syndrome (XH2 mutation) |
ARX mutations |
Testicular regression syndrome |
Defects in synthesis, secretion, or response to AMH |
Female genital ducts in otherwise normal men—“herniae uteri inguinale”; persistent müllerian duct syndrome |
Environmental chemicals (endocrine disrupters) |
Unclassified Forms of Disorders of Sexual Development |
In males |
Hypospadias |
Ambiguous external genitalia in 46,XY males with multiple congenital anomalies |
|
|
|
In females |
Absence or anomalous development of the vagina, uterus, and uterine tubes (Rokitansky-Küster syndrome) (WNT-4 mutation) |
* These terms are no longer recommended and should be abandoned. They are listed in parentheses during this transitional period.
** In pregnant patient with CAH whose disorder is poorly controlled or who is noncompliant especially during the first trimester.
AMH, anti-Müllerian hormone; ATRX, alpha-thalassemia X-linked mental retardation.
Data from Grumbach MM, Conte FA. Disorders of sex differentiation. In: Wilson JD, Foster DW, Kronenberg HM, Larsen PR, eds. Williams Textbook of Endocrinology. 9th ed. Philadelphia: W.B. Saunders; 1998:1303-1425.
At birth the external genitalia of affected girls are, as a rule, conspicuously abnormal, whereas the genitalia of affected boys are normally differentiated. The degree of masculinization can be judged by the size of the clitoris and the completeness of labioscrotal fusion, which determines the size of the urogenital sinus (Fig. 539-1). The phallus is invariably enlarged in the simple virilizing and salt-wasting forms, often approximating the size of a penis (eFig. 539.1  ). It is generally bound in chordee, behind which a perineal hypospadias is situated. In rare instances, the urethra extends to the tip of the phallus. The labia majora commonly looks like a bifid scrotum. Within the perineal opening of the urogenital sinus lie the orifices of the vagina and the urethra. Greater or lesser degrees of fusion of the labioscrotal folds produce a perineal opening that varies in size from that of a small urethra-like opening to a relatively normal female introitus with a separate urethra and vagina (see Fig. 539-1).
). It is generally bound in chordee, behind which a perineal hypospadias is situated. In rare instances, the urethra extends to the tip of the phallus. The labia majora commonly looks like a bifid scrotum. Within the perineal opening of the urogenital sinus lie the orifices of the vagina and the urethra. Greater or lesser degrees of fusion of the labioscrotal folds produce a perineal opening that varies in size from that of a small urethra-like opening to a relatively normal female introitus with a separate urethra and vagina (see Fig. 539-1).
The appearance of the external genitalia is not specific, and the genital abnormality can be indistinguishable from that in other forms of disordered sexual development with bilateral cryptorchidism. Measurement of plasma 17α-hydroxyprogesterone (17OHP) in the plasma is strikingly elevated in patients with 21-hydroxylase deficiency (both salt losers and non-salt-losers) as well as androstenedione and testosterone. Measurement of plasma 17OHP is the most useful diagnostic test, being used in neonatal screening programs for early diagnosis of this disorder (see Chapters 533 and 534).3-5
 OTHER FETAL SOURCES OF ANDROGEN
OTHER FETAL SOURCES OF ANDROGEN
3β-hydroxysteroid dehydrogenase (3HSD) is a rare disorder that affects both sexes, and female patients may have ambiguous genitalia. Familial glucocorticoid resistance is characterized by increased cortisol secretion without clinical evidence of hypercortisolism, but with manifestations of androgen and mineralocorticoid excess, caused by glucocorticoid receptor gene mutation, and rarely can lead to virilization in females.
 FETOPLACENTAL SOURCES OF ANDROGEN
FETOPLACENTAL SOURCES OF ANDROGEN
Placental aromatase has a critical role in protecting the female fetus and the mother from exposure to large amounts of testosterone synthesized mainly by the placenta.6-16 When deficient because of mutation in the P450arom gene (CYP19; locus 15q21-OMIM 107910), the concentration of androgen or androgen precursors exceeds the capacity for aromatization. As a consequence, the fetal placenta is unable to convert fetal adrenal androgen precursor to estrogen. This autosomal recessive disorder frequently results in virilization of the mother during pregnancy. The external genitalia may be ambiguous, or in the female there may be clitoromegaly. Wolffian duct derivatives are absent, and müllerian duct derivative are present. The ovaries may be multicystic in infancy and postpubertally. Typical features include hyper-gonadotropic hypogonadism, multicystic ovaries, osteopenia, delayed bone age, and tall stature after the age of puberty.17,18 Psychosocial orientation is normal, as is a response to estrogen therapy.
 PLACENTAL TRANSFER OF ANDROGENS FROM THE MOTHER
PLACENTAL TRANSFER OF ANDROGENS FROM THE MOTHER
In rare instances, a virilizing ovarian or adrenal tumor in the mother during pregnancy partially masculinizes its external genitalia. Therapeutic administration of medications with androgenic activity during pregnancy result in fusion of the labioscrotal folds and formation of a urogenital sinus if given before the 13th week of gestation, but enlargement of the clitoris can follow androgen treatment of the mother at any time during pregnancy.
 NONANDROGEN INDUCED XX, DISORDERS OF SEXUAL DEVELOPMENT
NONANDROGEN INDUCED XX, DISORDERS OF SEXUAL DEVELOPMENT
In these rare instances, accompanying developmental anomalies of the urinary tract and cloaca, often with atresia of the rectum or rectovaginal fistula, are observed.19 There also may be absence of a fallopian tube or an ovary, and the uterus may be poorly developed. Stenosis of the urethra can cause urinary retention in early infancy.
46,XY DISORDERS OF SEXUAL DEVELOPMENT (MALE PSEUDOHERMAPHRODITISM)
XY disorders of sexual development result from either the failure of the fetal testis to secrete adequate testosterone to bring about complete masculinization of the somatic sex structures or from defects in androgen-dependent target tissue response to released androgens (see Table 539-1). The karyotype is 46,XY, except in some patients with dysgenetic XY, DSD. Testes are present with variable degrees of ambisexual development of the genital ducts or the urogenital sinus and external genitalia, or both (Fig. 539-2). The appearance of the external genitalia varies from that of a normal girl to that of a boy with a penile urethra and either bilateral or unilateral cryptorchidism. Perineal hypospadias is common. The testes may be inside the abdomen, sometimes in the position of the ovaries, in the inguinal region, or in the labioscrotal folds.
 TESTICULAR UNRESPONSIVENESS TO hCG AND LH (LEYDIG CELL AGENESIS OR HYPOPLASIA)
TESTICULAR UNRESPONSIVENESS TO hCG AND LH (LEYDIG CELL AGENESIS OR HYPOPLASIA)
Autosomal recessive, homogenous, compound heterozygous mutations in the gene that encodes the hCG-LH receptor (LHGCR; 2p21), a G protein–coupled seven-transmembrane α-helical receptor, cause fetal and postnatal testosterone deficiency. The testes are small and undescended with absent or decreased numbers of Leydig cells. The external genitalia vary from female to ambiguous to hypoplastic male. Müllerian derivatives are absent.20-22 Plasma levels of 17-hydroxyprogesterone, androstenedione, and testosterone are low, and stimulation with hCG evokes little or no increase. Basal FSH and LH levels are elevated in infancy and at puberty.
 INBORN ERRORS OF TESTOSTERONE BIOSYNTHESIS
INBORN ERRORS OF TESTOSTERONE BIOSYNTHESIS
Figure 531-1 shows the major pathways in testosterone biosynthesis. Each step is associated with an enzymatic defect inherited as an autosomal recessive trait that causes incomplete masculinization of the urogenital sinus or external genitalia (Table 539-1) but not differentiation of müllerian duct structures. The disorders that affect synthesis of corticosteroids and androgens include 3β-hydroxysteroid dehydrogenase-2 deficiency, P450c17 (17α-hydroxylase) deficiency, P450 oxidoreductase deficiency, and Δ7-sterol reductase deficiency (Smith-Lemli-Opitz syndrome), are discussed in more detail in Chapter 533. Enzyme deficiencies that affect testosterone synthesis but not corticosteroid synthesis include 17,20 lyase (P450C17) deficiency and 17β-hydroxysteroid dehydrogenase (HSD 17-β3) type 3 deficiency. In congenital lipoid hyperplasia (StAR deficiency), the Leydig cells are destroyed early in gestation, eliminating testosterone biosynthesis.5 The patients have enormous accumulations of lipid in the cells of the adrenal cortex but surprisingly less in the fetal testis, severe adrenal insufficiency, and death in early infancy if not treated.23,24 Boys have ambiguous external genitalia, a blind vaginal pouch, and undescended testes, and with a severe defect, female external genitalia. The enlarged adrenal glands (with rare exceptions) displace the kidneys caudally. Girls have a normal phenotype but also have life-threatening salt loss and cortisol deficiency. Girls feminize normally at puberty because lipids have not accumulated and destroyed the cells before the pubertal increase in secretion of gonadotropins. In P450 side chain cleavage deficiency genitalia are female, rarely ambiguous. Wolffian duct derivatives are hypoplastic or normal, müllerian duct derivatives are absent. Concomittant adrenal insufficiency is either severe in infancy or milder with onset in childhood. Individuals with P450 oxidoreductase (POR) deficiency have testes with a spectrum of genital abnormalities from ambiguous to hypospadias to normal males. Wolffian duct derivatives are absent or hypoplastic. Features of Antley-Bixler syndrome (craniostenosis, choanalatresia/stenosis, multiple skeletal malformations) are seen in 30% to 65% of individuals with this mutation. 46,XY individuals with 3 β-hydroxysteroid dehydrogenase type II Δ5isomerase deficiency (3βHSD2) have testes with ambiguous external genitalia, or hypospadia, normal wolffian duct derivatives, severe adrenal insufficiency in infancy, poor virilization at puberty with gynecomastia. Patients with CYP17 mutations have impaired synthesis of 17OH-progesterone, 17α-hydroxypregnenolone and their products (androgens, estrogens, and cortisol) caused by an autosomal-recessive mutant gene.25-28 The gene is located on chromosome 10q24.3 and encodes both the adrenal and testicular enzymes that catalyze 17-hydroxylation of pregnenolone and progesterone and removal of the C21 side chain of 17-hydroxypregnenolone (C17-20-lyase) to form C19 steroid DHEA.29 Increased secretion of corticosterone and deoxycorticosterone causes hypokalemic alkalosis and low-renin hypertension. Plasma concentration of gonadal steroids and excretion of urinary 17-ketosteroids are low. Affected boys are incompletely masculinized with hypospadias or may appear to be phenotypic girls, with a blind vaginal. Virilization is poor at puberty and gynecomastia is common.
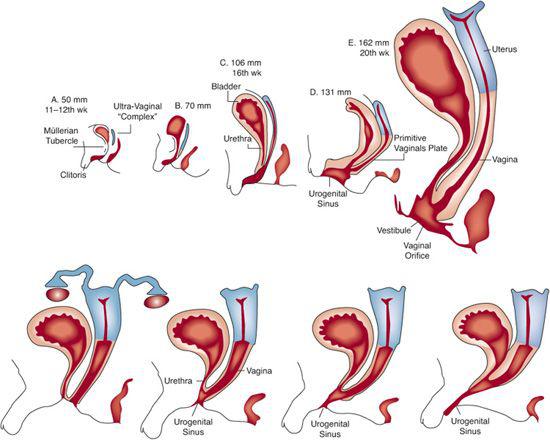
FIGURE 539-1. Development of XX DSD. Top: Sequence of differentiation of female accessory sex structures. Gradual descent of uterovaginal complex is evident. To modify differentiation of the urogenital sinus, especially the urethral groove, it seems that androgens must act on the female fetus before the 13th week of gestation, although enlargement of the clitoris can be induced at later stages. Bottom: Variations in degree of masculinization of urogenital sinus and external genitalia in androgen-induced XX DSD.
Individuals with 17,20 lyase (P450C17) deficiency have ambiguous external genitalia and inguinal or intra-abdominal testes. At puberty, incomplete masculinization can occur. Gynecomastia is infrequent. If the disorder is diagnosed in infancy, these patients can be reared as boys and treated with testosterone to induce male secondary characteristics and phallic growth. Plasma levels of testosterone, androstenedione, estradiol, and DHEA are low. After stimulation with hCG, the ratio of C21 steroids (cortisol, 17OHP) to C19 steroids increases. This disorder must be differentiated from 5-reductase-2 deficiency and from 17βHSD-3 deficiency. 46,XY individuals with mutations in the gene encoding 17β-hydroxysteroid dehydrogenase type 3 (or 17-ketosteroid reductase)29-33 have female or ambiguous external genitalia, inguinal testes, male genital duct development, and progressive virilization at puberty, usually with concomitant breast development. The type 3 isozyme, the last enzyme in the biosynthetic pathway for testosterone, is expressed primarily in the testis. Levels of plasma androstenedione and estrone are strikingly elevated because their conversion to testosterone and estradiol is impaired. These patients must be differentiated from those with partial (incomplete) androgen insensitivity syndrome or 5α-reductase-2 deficiency, who have a similar phenotype but not a similar sex steroid pattern.
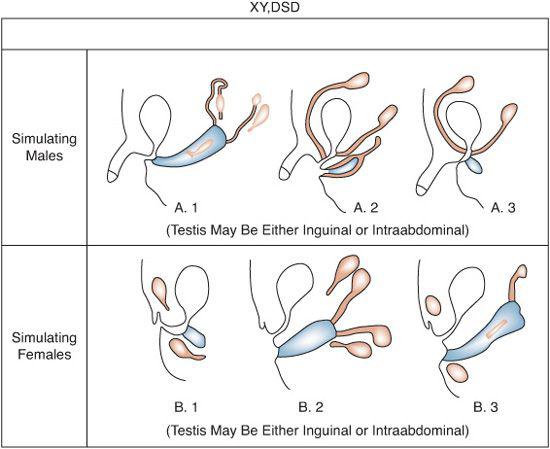
FIGURE 539-2. Common anatomic findings in XY, DSD. Black structures are testes, derivatives of wolffian ducts. Cross-hatched areas include derivatives of müllerian ducts and female urogenital structures.
 ANDROGEN RECEPTOR DEFECTS
ANDROGEN RECEPTOR DEFECTS
The syndrome of complete androgen insensitivity results from a mutation in the X-linked androgen receptor, a ligand-regulated transcription factor that binds both dihydrotestosterone and testosterone (eFig. 539.2  ). It is estimated to occur in between 1:20,000 and 1:60,000 live male births. Family pedigrees show X-linked inheritance consistent with the location of the androgen receptor gene on Xq11-q12. In addition to transmission from the mother by the mutant gene on the X-chromosome of her oocyte, it is estimated that in about 30% of these individuals the mutation occurs in the postzygotic stage and leads to somatic mosaicism.34-40 The testes secrete testosterone, and at puberty the concentration of plasma testosterone usually is within the normal range for boys. However, both genital and somatic end organs do not respond to androgens in the fetus or at puberty, which leads to female differentiation of the urogenital sinus and external genitalia and to feminization at puberty. The hypothalamic feedback mechanism lacks normal sensitivity to testosterone, which elevates serum levels of luteinizing hormone (LH), but the concentration of follicle-stimulating hormone (FSH) in the serum usually is normal because of circulating inhibin-B and estradiol feedback. These individuals have a normal female appearance so the diagnosis is often not suspected. The testes usually are in the inguinal canal or in the labial folds. In some instances, the clitoris is slightly enlarged and the labioscrotal folds are partially fused. There is a characteristic blind vaginal pouch, absent (or rudimentary, in about 30%) müllerian duct derivatives due to the action of antimüllerian hormone by the fetal testis, and absent or hypoplastic wolffian ducts.41 Development of the genital ducts is variable, but the uterus is absent.
). It is estimated to occur in between 1:20,000 and 1:60,000 live male births. Family pedigrees show X-linked inheritance consistent with the location of the androgen receptor gene on Xq11-q12. In addition to transmission from the mother by the mutant gene on the X-chromosome of her oocyte, it is estimated that in about 30% of these individuals the mutation occurs in the postzygotic stage and leads to somatic mosaicism.34-40 The testes secrete testosterone, and at puberty the concentration of plasma testosterone usually is within the normal range for boys. However, both genital and somatic end organs do not respond to androgens in the fetus or at puberty, which leads to female differentiation of the urogenital sinus and external genitalia and to feminization at puberty. The hypothalamic feedback mechanism lacks normal sensitivity to testosterone, which elevates serum levels of luteinizing hormone (LH), but the concentration of follicle-stimulating hormone (FSH) in the serum usually is normal because of circulating inhibin-B and estradiol feedback. These individuals have a normal female appearance so the diagnosis is often not suspected. The testes usually are in the inguinal canal or in the labial folds. In some instances, the clitoris is slightly enlarged and the labioscrotal folds are partially fused. There is a characteristic blind vaginal pouch, absent (or rudimentary, in about 30%) müllerian duct derivatives due to the action of antimüllerian hormone by the fetal testis, and absent or hypoplastic wolffian ducts.41 Development of the genital ducts is variable, but the uterus is absent.
At puberty, estradiol secreted by the testes and synthesized in extragonadal tissues by the aromatization of testosterone bring about feminization of body habitus, development of breasts, and estrogenization of the vaginal mucosa despite elevated testosterone levels, but menstruation does not occur. Mean height is taller than average for women. The discovery of a testis in an inguinal or labial hernia usually is the only clue to the diagnosis in the absence of a familial history. About 1% of females with bilateral inguinal herniae (uncommon in girls) have congenital androgen insensitivity syndrome. The diagnosis is considered when an adolescent girl has primary amenorrhea (it is the third most common cause after gonadal dysgenesis and congenital absence of the vagina) in the presence of otherwise female secondary sexual characteristics (including breast development), especially when associated with absent or sparse sexual hair and unilateral or bilateral hernial masses. Among most patients, pubic and axillary hair are absent or sparse, a manifestation of the impaired response to androgen of the hair follicles that give rise to sexual hair. In the classic form of the syndrome, large amounts of testosterone or dihydrotestosterone do not induce either masculinization or an appropriate degree of protein anabolism. Castration causes a decrease in plasma levels of estradiol and testosterone, an increase in both FSH and LH levels, and menopausal symptoms. There is a low risk for the testes to undergo neoplastic transformation before adulthood. Orchiectomy is recommended in late adolescence or early adulthood after puberty is complete and should be followed by estrogen replacement therapy.
Some patients have a gene mutation that encodes a defective but partially functional androgen receptor—the partial androgen insensitivity syndrome.39,42-44 These individuals have breast development at puberty and undergo variable degrees of masculinization. There is clitoral enlargement at birth, and some patients have ambiguous external genitalia. Phenotypic males with infertility as the sole manifestation of a deficiency of androgen receptors have been described. They represent one extreme of the highly variable phenotype of androgen resistance.39
 5α-REDUCTASE TYPE 2 DEFICIENCY (PSEUDOVAGINAL PERINEOSCROTAL HYPOSPADIAS
5α-REDUCTASE TYPE 2 DEFICIENCY (PSEUDOVAGINAL PERINEOSCROTAL HYPOSPADIAS
Patients with this syndrome have an XY karyo-type but have impaired enzymatic transformation of testosterone to its active hormone (dihydrotestosterone) at testosterone sensitive end organs. The disorder is autosomal recessive and caused by mutations in the 5α-reductase type 2 gene (5RD5A2) on chromosome 2p23. The testes are normally differentiated, internal genital ducts are male, but the external genitalia are ambiguous.45-47 At birth, the phallus usually is small and hypospadiac. There is persistence of the urogenital sinus with a blind vaginal pouch. In severe disease, separate vaginal and urethral orifices are present. At puberty type 1-5α-reductase isoenzyme is expressed (this enzyme is not expressed in the fetus), leading to masculinization including phallus enlargement, growth of axillary and pubic hair, and in many children, male gender identity.48 Acne, facial hair, and temporal recession of the hairline are minimal or absent. Unlike patients with androgen receptor defects, these patients do not have gynecomastia. The diagnosis is suggested by the finding of an abnormally high plasma testosterone to dihydrotestosterone ratio (> 8.5 in infancy) after administration of hCG (1500 IU/m2 intramuscularly daily × 3 doses or every 48 hours × 7 doses with blood sampling 24 hours after the last dose). It is confirmed by assay of 5α-reductase in cultures of skin fibroblasts, detection of an increased 5β/5α ratio of urinary C19 and C21 steroid metabolites, and preferably by DNA analysis of the 5α-reductase type 2 gene. Early diagnosis is critical because selection of male sex assignment necessitates testosterone or dihydrotestosterone therapy to induce growth of the phallus. Repair of hypospadias is performed in early childhood.
 DYSGENETIC XY, DISORDERS OF SEXUAL DEVELOPMENT
DYSGENETIC XY, DISORDERS OF SEXUAL DEVELOPMENT
Defective testicular organogenesis results in ambiguous development of the genital ducts, urogenital sinus, and external genitalia. A heterogeneous group of disorders, including 45,X/46,XY mosaicism, structural abnormalities of the Y chromosome, deletions and mutations of the testicular determiner gene (SRY), mutations in autosomal genes, duplication of DSS (dosage sensitive sex) locus on the X chromosome, and the sporadic and heritable forms of XY gonadal dysgenesis manifest defective gonadogenesis and masculinization.
A highly diverse phenotype has been described among patients with XO/XY mosaicism or structural abnormality of the Y chromosome. The appearance can range from a sexually infantile female phenotype with or without the somatic anomalies of Turner syndrome and with bilateral streak gonads, through variable degrees of masculine differentiation of the external genitalia, urogenital sinus, and genital ducts, to almost normal male differentiation of the genital tract.20,48,49 Some patients have a dysgenetic testis on one side and a streak gonad on the other. Short stature and the somatic anomalies of Turner syndrome are inconstant features. The dysgenetic testes must be removed because of the increased tendency toward development of malignant tumors. Patients with an incomplete form of familial XY gonadal dysgenesis are of normal stature and do not have the components of Turner syndrome. The testes are dysgenetic, and there are usually both müllerian and wolffian duct derivatives. The external genitalia are ambiguous. The gonads have variable degrees of dysgenetic testicular differentiation, and some virilization occurs at puberty. Gonadotropin levels are elevated. The defective testes are removed because of the risk of neoplastic transformation. Familial XY gonadal dysgenesis may be transmitted as an X-linked recessive or sex-limited autosomal dominant trait, or owing to a mutant SRY gene. Both the complete form, which has a female phenotype, and the incomplete form can occur in the same pedigree.
A variety of gene defects are associated with XY, disorders of sexual development. These include 3 syndromes caused by mutations of the Wilms tumor suppressor gene (WT1).50-54Denys-Drash syndrome50 is associated with degenerative renal disease (focal and diffuse mesangial sclerosis), early onset nephrotic syndrome with rapid progression to end-stage renal disease, hypertension, and commonly with Wilms tumor at a median age of 18 months. Both the testes and the kidneys are dysgenetic and predisposed to malignant transformation. In Frasier syndrome,51,52 individuals have female or ambiguous external genitalia, later-onset nephropathy (focal and segmental glomerulosclerosis), and a predisposition for gonadoblastoma but not Wilms tumor. In Wilms tumor–aniridia–gonadoblastoma–mental retardation (WAGR) contiguous gene deletion (WT1) syndrome in region chromosome 11p13, affected boys often have ambiguous or hypoplastic male genitalia. Heterozygous mutations in the SOX-9 (SRY-related high-mobility group box) gene are associated with a severe bone dysmorphology syndrome, camptomelic dysplasia, and frequently dysgenetic XY, disorders of sexual development. Other gene mutations associated with disorders of sexual development include desert hedgehog (DHH), TSPYL1 (associated with sudden death in infancy). ARX (associated with X-linked lissencephaly), duplication of the X-linked DAX1 (AHC),60 9p-syndrome (DMRT1); DHCR7 (Smith-Lemli-Opitz syndrome), and XH2 (the ATR-X syndrome: X-linked -thalassemia, mental retardation, small testes, and ambiguous external genitalia.55-61
 DEFECTS IN SYNTHESIS, SECRETION, OR RESPONSE TO ANTIMULLERIAN HORMONE
DEFECTS IN SYNTHESIS, SECRETION, OR RESPONSE TO ANTIMULLERIAN HORMONE
These disorders result in the persistent müllerian duct syndrome characterized by normal male development of the external genitalia, testes, normal male ducts, and müllerian duct derivatives.62,63 The diagnosis often is made by the finding of a fallopian tube and uterus in a patient undergoing inguinal hernia repair, orchiopexy, or abdominal surgery. The disorder is inherited as a sex-limited autosomal-recessive trait and is caused by mutations in the gene encoding the antimüllerian hormone or its receptor (AMHR2). In the former (in contrast to the latter), the plasma concentration of AMH is low or not detected.
 OVOTESTICULAR DISORDERS OF SEXUAL DEVELOPMENT (TRUE HERMAPHRODISM)
OVOTESTICULAR DISORDERS OF SEXUAL DEVELOPMENT (TRUE HERMAPHRODISM)
These persons have both an ovary and a testis or, more commonly, one or both gonads are ovotestes.85 Development of the accessory sexual structures is highly variable. Three-fourths of these patients have been reared as boys, but they have variable degrees of hypospadias. Cryptorchidism and an inguinal hernia that contains a gonad or vestigial uterus and fallopian tube are present in 50% of patients. Predominantly masculine or feminine maturation occurs at puberty. Most patients do not have a sex chromosome abnormality.  When the sex of rearing is chosen, an important aim is to preserve the appropriate gonad or gonadal segment, especially if an ovary is present in the mesosalpinx or a testis is attached to its exocrine ducts in the scrotum. There is, however, an increased risk of gonadal neoplasia.
When the sex of rearing is chosen, an important aim is to preserve the appropriate gonad or gonadal segment, especially if an ovary is present in the mesosalpinx or a testis is attached to its exocrine ducts in the scrotum. There is, however, an increased risk of gonadal neoplasia.
 ENVIRONMENTAL CHEMICALS
ENVIRONMENTAL CHEMICALS
An increase in the prevalence of hypospadias, cryptorchidism, cancer of the testes, and in some European countries a fall in the sperm count has occurred over the past 30 years.66-104 It has been speculated that the increased prevalence is related to exposure in utero and postnatally to environmental endocrine disrupters, especially compounds with estrogenic activity and agents that bind to the fetal androgen receptors; for example, phthates as well as herbicides and fungicides inhibit androgen action. Further studies on the levels and risks of natural and environmental estrogens and antiandrogens in humans are necessary before the putative increase in prevalence of these abnormalities including the testicular dysgenesis syndrome can be attributed to these agents.
UNCLASSIFIED FORMS OF ABNORMAL SEX DIFFERENTIATION
The underlying cause of several abnormalities of sex differentiation is unknown; therefore, they remain categorized as unclassified (see Table 539-1). For example, ambiguous external genitalia are associated with a large number of malformation syndromes such as Aarskog-Scott, CHARGE, and VATER syndromes.66,67
SYNDROMES OF GONADAL DYSGENESIS AND VARIANTS
 TURNER SYNDROME
TURNER SYNDROME
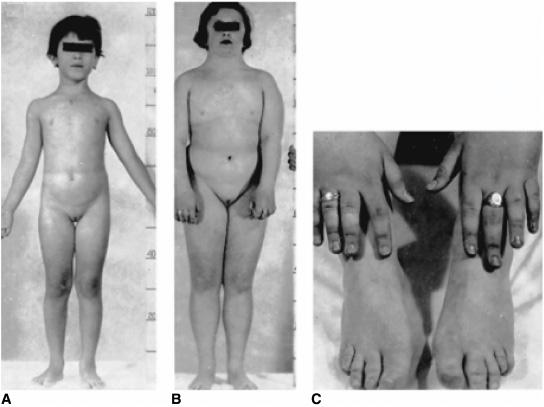
Haploinsufficiency of the X chromosome (45,XO karyotype) results in a female phenotype with associated findings of short stature, sexual infantilism, streak gonads, and a diversity of associated somatic anomalies. It is estimated that about 2% of all zygotes are 45,XO (probably the most common chromosome anomaly in the human), but less than 0.2% of 45,X fetuses survive to term such that about 7% of spontaneous abortions have a 45,X karyotype. The incidence of Turner syndrome is about 1 per 2000 live phenotypic female births; approximately 50% have a 45,X karyotype. Intrauterine growth retardation is common. Maternal age is not advanced. There is increased prevalence of twinning, but familial instances are exceedingly rare. Lymphedema and loose folds of skin around the nape of the neck (Bonnevie-Ullrich syndrome) suggest the diagnosis during the neonatal period (Fig. 539-3) which is then confirmed by genetic testing. Pleural effusion may occur among newborns.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


