Disorders of Purine and Pyrimidine Metabolism
Georges van den Berghe
PURINE METABOLISM
 METABOLIC PATHWAYS
METABOLIC PATHWAYS
Purines comprise bases, nucleosides in association with ribose or deoxyribose, and nucleotides with one or more added phosphate groups. Purine nucleotides are essential cellular constituents. They are the building blocks of the polynucleotides, DNA and RNA, and, under the form of mononucleotides or of nucleosides, also intervene in numerous cellular functions. Among these are energy transfer (eg, by adenosine triphosphate [ATP]), metabolic regulation (eg, by guanosine triphosphate [GTP]), and signaling (eg, by adenosine). Purine metabolism can be divided into three pathways (Fig. 168-1):
1. The biosynthetic pathway, starts with the formation, often termed de novo, of the high-energy compound phosphoribosyl pyrophosphate (PRPP) and leads in 10 steps to the synthesis of the nucleoside monophosphate inosine monophosphate (IMP). From IMP, two reactions lead to the formation of adenosine monophosphate (AMP). Subsequently, the nucleoside di- and triphosphates ADP and ATP and their deoxy counterparts are synthesized. Two other reactions convert IMP into GMP, from which GDP, GTP, and their deoxy counterparts are formed.
2. The purine catabolic pathway starts from the nucleoside monophosphates GMP, IMP, and AMP and produces uric acid, a poorly soluble compound that tends to crystallize once its plasma concentration increases above 6.5 to 7.0 mg/dl (0.38–0.47 mmol/l).
3. The purine salvage pathway utilizes the purine bases guanine, hypoxanthine, and adenine, which are provided by food intake or the catabolic pathway, and reconverts them by phosphoribosylation into GMP, IMP, and AMP, respectively. It also utilizes the purine nucleosides adenosine, guanosine, and their deoxy counterparts by phosphorylation into the corresponding monophosphates, catalyzed by kinases.
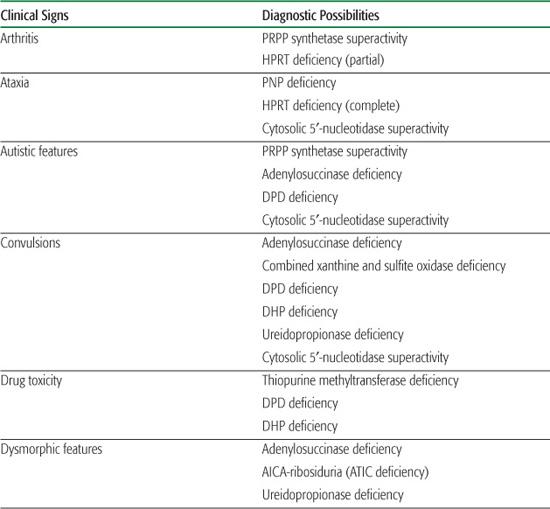
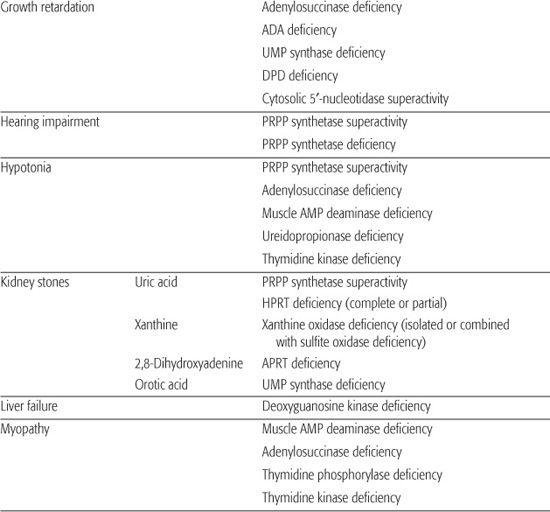
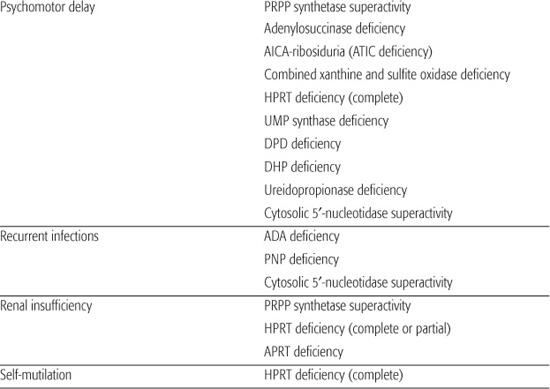
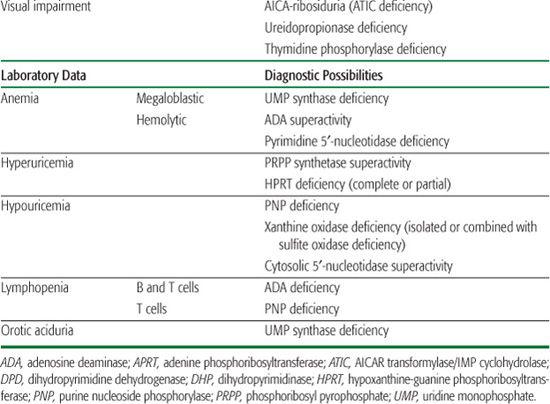
Inborn errors of purine metabolism comprise errors of purine nucleotide synthesis, of purine catabolism, and of purine salvage. They should be considered in patients with hyper- or hypouricemia, kidney stones, and a variety of muscle, neurological, and other symptoms (Table 168.1). The deficiencies of adenosine deaminase and purine nucleoside phosphorylase, two purine catabolic enzymes, cause severe combined immunodeficiency (SCID; see Chapter 188). Adenosine deaminase superactivity causes hemolytic anemia. The deficiency of deoxyguanosine kinase causes a mitochondrial disease. The deficiency of thiopurine methyltransferase is of importance in pharmacogenetics.
DISORDERS OF DE NOVO PURINE SYNTHESIS
 PHOSPHORIBOSYLPYROPHOSPHATE SYNTHETASE SUPERACTIVITY
PHOSPHORIBOSYLPYROPHOSPHATE SYNTHETASE SUPERACTIVITY
Phosphoribosylpyrophosphate (PRPP) synthetase superactivity (OMIM 311850), one of the few known examples of a hereditary anomaly that enhances the activity of an enzyme, was initially reported in a patient with early adult-onset gout. Since then, about two dozen families have been identified in which excessive production of uric acid could be traced to this disorder.2,3
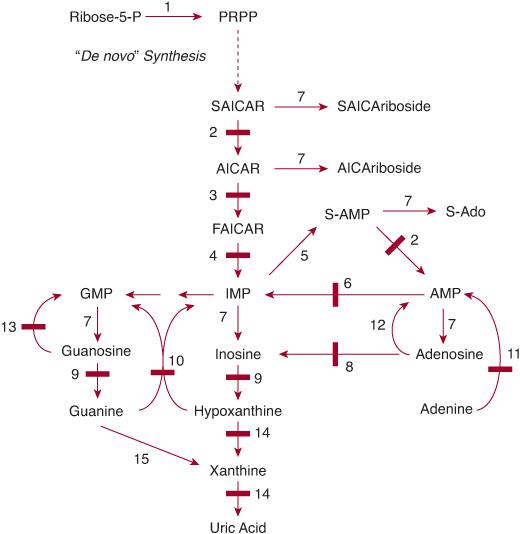
FIGURE 168-1. Pathways of purine metabolism. (1) PRPP (phosphoribosylpyrophosphate) synthetase; (2) adenylosuccinate lyase (adenylosuccinase); (3) AICAR transformylase; (4) IMP cyclohydrolase (3 and 4 form ATIC); (5) adenylosuccinate synthetase; (6) AMP deaminase; (7) 5′-nucleotidase(s); (8) adenosine deaminase; (9) purine nucleoside phosphorylase; (10) hypoxanthine-guanine phosphoribosyltransferase; (11) adenine phosphoribosyltransferase; (12) adenosine kinase; (13) guanosine kinase; (14) xanthine oxidase (dehydrogenase); (15) guanine deaminase. Enzyme defects are indicated by solid bars. AICAR, aminoimidazolecarboxamide ribotide; AMP, adenosine monophosphate; ATIC, 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase; FAICAR, formylaminoimidazolecarboxamide ribotide; GMP, guanosine monophosphate; IMP, inosine monophosphate; P, phosphate; PRPP, phosphoribosyl pyrophosphate; S-Ado, succinyladenosine; SAICAR, succinylaminoimidazolecarboxamide ribotide; S-AMP, adenylosuccinate; XMP, xanthosine monophosphate.
This extremely rare, X-linked, gain-of-function enzyme anomaly causes hyperuricemia and hyperuricosuria in young adult males, which may lead to uric acid lithiasis, gouty arthritis, and renal insufficiency. In some patients, symptoms already appear in infancy and are accompanied by sensorineural deafness, hypotonia, ataxia, and autistic features that may also be found in heterozygous females. The increased enzyme activity results in enhanced generation of PRPP and hence of de novo synthesis of uric acid. Treatment with allopurinol, which inhibits xanthine oxidase, decreases production of uric acid. To prevent stone formation, high fluid intake is recommended and urine alkalinized.
Recently, loss-of-function mutations of a PRPP synthetase gene have been identified in Arts syndrome, Rosenberg-Chutorian syndrome, and Charcot-Marie-Tooth disease CMTX5, characterized by peripheral neuropathy, hearing impairment, and optic atrophy. 
 ADENYLOSUCCINATE LYASE DEFICIENCY
ADENYLOSUCCINATE LYASE DEFICIENCY
The deficiency of adenylosuccinate lyase (ADSL, also known as adenylosuccinase; OMIM 103050) is the first enzyme defect reported in humans to affect the de novo pathway of purine synthesis.12 Over 60 patients have been identified so far.13,14
Clinical presentation of this enzyme defect is widely variable, including severe neonatal seizures, mild to profound mental retardation, and autistic features. The hallmark of the disorder is the accumulation of succinylaminoimidazole carboxamide riboside and succinyladenosine in CSF and urine. These are the products of the dephosphorylation of the two substrates of the enzyme, which intervenes twice in purine synthesis (Fig. 168-1). The disorder is autosomal recessive, and a variety of disease-causing mutations have been identified in more than 50 patients. Treatment with adenine and allopurinol and with ribose have been tried with little success. 
 AICA-RIBOSIDURIA (ATIC DEFICIENCY)
AICA-RIBOSIDURIA (ATIC DEFICIENCY)
In a female infant with profound mental retardation, dysmorphic features (prominent forehead, brachycephaly, wide mouth, low-set ears, prominent clitoris), and blindness, the following were identified: a massive excretion of AICA-riboside, the dephosphorylated counterpart of AICAR (Fig. 168-1); a deficiency of ATIC, the bifunctional enzyme catalyzing the two last steps of purine biosynthesis; and lesions of its gene. 
Table 168-1. Main Presenting Clinical Signs and Laboratory Data in Inborn Errors of Purine and Pyrimidine Metabolism
DISORDERS OF PURINE CATABOLISM
 MYOADENYLATE DEAMINASE DEFICIENCY (MUSCLE AMP DEAMINASE DEFICIENCY)
MYOADENYLATE DEAMINASE DEFICIENCY (MUSCLE AMP DEAMINASE DEFICIENCY)
The deficiency of myoadenylate deaminase (OMIM 102770), the muscle-specific isoenzyme of AMP deaminase (AMPD), was first reported by Fishbein and colleagues.42 Since then, the defect has been found in 1% to 2% of the Caucasian population, but most deficient individuals are asymptomatic.43 In some subjects, it is manifested by muscle weakness and cramping following vigorous exercise. It is usually diagnosed in adulthood but may be a common cause of benign hypotonia or cardiomyopathy in infancy. The disorder is transmitted as an autosomal recessive trait. Most affected individuals carry the same nonsense mutation resulting in a stop codon. Patients should be advised to exercise with caution. Administering ribose has been reported to improve muscle strength and endurance. 
 ADENOSINE DEAMINASE DEFICIENCY
ADENOSINE DEAMINASE DEFICIENCY
Adenosine deaminase (ADA) catalyzes the deamination of adenosine and its deoxy counterpart deoxyadenosine. A gross deficiency of ADA was first reported by Giblett and colleagues58 in two unrelated children with profound impairment of both cellular (T-cell) and humoral (B-cell) immunity, known as severe combined immunodeficiency disease (SCID). Since that time, several hundred patients with ADA deficiency have been diagnosed. The disorder is discussed in Chapter 188.
 ADENOSINE DEAMINASE SUPERACTIVITY
ADENOSINE DEAMINASE SUPERACTIVITY
A hereditary, approximately fiftyfold elevation of erythrocyte ADA has been shown to cause nonspherocytic hemolytic anemia.59 It is explained by an enhanced catabolism of the adenine nucleotides, including ATP, owing to the increased activity of ADA.
 PURINE NUCLEOSIDE PHOSPHORYLASE DEFICIENCY
PURINE NUCLEOSIDE PHOSPHORYLASE DEFICIENCY
Their finding of ADA deficiency prompted Giblett and colleagues to search for other defects of purine and pyrimidine metabolism in patients with immune disorders. This resulted in the discovery of purine nucleoside phosphorylase (PNP) deficiency in a child with an isolated defect of T-cell function.60 The disorder is much less frequent than ADA deficiency, with about 50 patients reported. It is discussed in Chapter 188.
 XANTHINE OXIDASE DEFICIENCY
XANTHINE OXIDASE DEFICIENCY
The deficiency of xanthine oxidase (also termed xanthine dehydrogenase and xanthine oxidoreductase, or XOR), characterized by xanthinuria, was the first enzyme defect reported in human purine metabolism.61 Originally described as a benign disorder, it was subsequently found that it could also occur in combination with a devastating disorder similar to isolated sulfite oxidase deficiency.62 The combination is caused by inability to synthesize a molybdenum cofactor common to three oxidases: XOR, aldehyde oxidase, and sulfite oxidase. Accordingly, three types of XOR deficiency are now recognized: (1) type I, isolated XOR deficiency (OMIM 278300); (2) type II, combined XOR and aldehyde oxidase deficiency (OMIM 603592), which causes equally benign xanthinuria; and (3) combined deficiency of XOR, aldehyde, and sulfite oxidase (OMIM 252150), provoking a lethal disease.
Isolated xanthine oxidase deficiency is asymptomatic in more than half of the cases, which can be identified by low serum uric acid levels and increased urinary xanthine. Symptomatic individuals develop brownish yellow, radiolucent xanthine stones that can lead to urinary tract complications and even chronic renal failure. Inheritance is autosomal recessive. In order to prevent renal stones, a low purine diet and high fluid intake should be prescribed.
Xanthine oxidase needs a molybdenum cofactor, which is also required for sulfite oxidase. Defects of this cofactor cause a combined defect of both enzymes in which the clinical picture of sulfite oxidase deficiency (which is also found as an isolated defect; see Chapter 157) dominates. Symptoms include intractable seizures and severe mental retardation. 
DISORDERS OF PURINE SALVAGE
 HYPOXANTHINE-GUANINE PHOSPHORIBOSYLTRANSFERASE DEFICIENCY
HYPOXANTHINE-GUANINE PHOSPHORIBOSYLTRANSFERASE DEFICIENCY
In 1967, Seegmiller and coworkers discovered a complete deficiency of hypoxanthineguanine phosphoribosyltransferase (HPRT or HGPRT) in children with the Lesch-Nyhan syndrome (OMIM 300322), an incapacitating neurological disorder that, with few exceptions, is limited to males.69 It is characterized by choreoathetosis, spasticity, compulsive self-mutilation manifested by biting away lips and tongue and ends of the fingers, and three- to fourfold normal uric acid levels in plasma and urine.70 That same year, a partial deficiency of HPRT was described in patients with milder presentations, sometimes termed Kelley-Seegmiller syndrome or now more often called Lesch-Nyhan variants (OMIM 300323); this disorder involves overproduction of uric acid that leads to severe gouty arthritis in early adult life and involves minor or no neurological dysfunction.71 This led to the identification of a continuous spectrum of HPRT deficiencies, ranging from gout to a devastating neurological syndrome.72
When profound, HPRT deficiency typically manifests at 6 to 10 months of age with spasticity, choreiform movements, compulsory self-mutilation, and mental retardation. Individuals with a partial deficiency of the enzyme have hyperuricemia and gout without neurological manifestations. The hyperuricemia is explained by the increased availability of PRPP, resulting in an enhancement of de novo purine synthesis. The neurological symptoms might result from dopaminergic dysfunction. The disorder is X-linked, and over 250 mutations of the HPRT gene have been identified. Treatment with allopurinol, which inhibits xanthine oxidase, decreases the overproduction of uric acid but has no effect on the neurological symptoms. 
 ADENINE PHOSPHORIBOSYLTRANSFERASE DEFICIENCY
ADENINE PHOSPHORIBOSYLTRANSFERASE DEFICIENCY
The deficiency of adenine phosphoribosyltransferase (APRT; OMIM 102600) was first identified in heterozygotes with a partial enzyme defect.106 Later, Cartier and Hamet107 reported a homozygous 4-year-old with urinary crystals and stones. Approximately 300 patients have been diagnosed worldwide, but up to 50% of APRT-deficient subjects may be asymptomatic.
Adenine phosphoribosyltransferase (APRT) deficiency may cause urinary passage of gravel, radiolucent crystals, or small stones often accompanied by renal colic and infections and even anuric renal failure. The deficiency results in the loss of adenine salvage that is then oxidized by xanthine oxidase into 2,8-dihydroxyadenine, a compound that is several-fold less soluble than uric acid. Inheritance is autosomal recessive. Patients may have no detectable enzyme activity or a partial deficiency; this is quite common in Japan. Treatment comprises allopurinol to inhibit xanthine oxidase, purine restriction, and high fluid intake. 
 DEOXYGUANOSINE KINASE DEFICIENCY
DEOXYGUANOSINE KINASE DEFICIENCY
In three large consanguineous kindreds of Druze origin with the hepatocerebral form of mitochondrial DNA depletion syndrome (characterized by early progressive liver failure, neurological abnormalities, hypoglycemia, and increased lactate), homozygosity mapping was used to search for the causative gene defect.113 It led to the identification of a mitochondrial deoxyguanosine kinase deficiency. This enzyme phosphorylates the deoxy counterpart of guanosine (Fig. 168-1) into deoxy GMP. The disorder is discussed in Chapter 176.
 THIOPURINE METHYLTRANSFERASE DEFICIENCY
THIOPURINE METHYLTRANSFERASE DEFICIENCY
Thiopurine S-methyltransferase (TPMT) catalyzes the S-methylation of several synthetic pharmacological purine analogs that contain a thiol group such as 6-mercaptopurine, 6-thioguanine, and azathioprine that is converted to 6-mercaptopurine in vivo. These drugs are used to treat various diseases, including cancers, rheumatoid arthritis, and other autoimmune disorders, and immunosuppressants after organ transplantation. They are converted via phosphoribosylation by hypoxanthine-guanine phosphoribosyltransferase and adenine phosphoribosyltransferase into active thionucleotides, which exert their therapeutic action by incorporation into DNA and RNA. Their oxidation by xanthine oxidase and S-methylation by TPMT results in inactivation.
The wide variations in therapeutic response and occurrence of toxic side effects in some patients receiving thiopurines led to the identification of TPMT as a determining factor in this variability.114,115 Approximately 90% of individuals in various ethnic populations have high TPMT activity, about 10% have intermediate activity, and 1 in 300 lack activity. Patients with no or less efficient methylation of thiopurines have more extensive conversion to active thionucleotides that leads to severe, potentially fatal myelosuppression. Therefore, determining the TPMT status prior to treatment with thiopurines is now recommended in predictive pharmacogenetics.116
PYRIMIDINE METABOLISM
 METABOLIC PATHWAYS
METABOLIC PATHWAYS
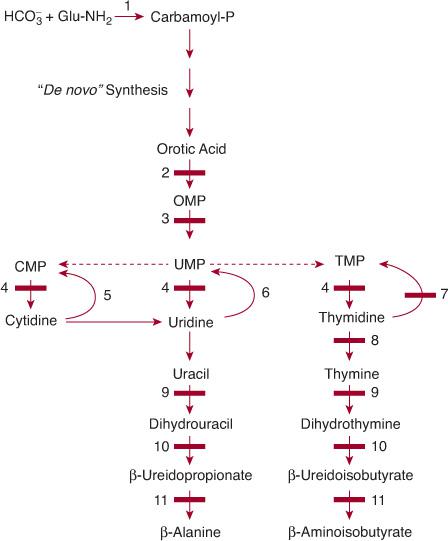
Similar to that of the purine nucleotides, the metabolism of the pyrimidine nucleotides can be divided into three pathways (Fig. 168-3):
1. The biosynthetic pathway, also often termed de novo, starts with the formation of the high-energy compound carbamoyl phosphate by cytosolic carbamoyl phosphate synthetase (CPS II). This enzyme is different from mitochondrial CPS I, which catalyzes the first step of ureogenesis. Formation of carbamoyl phosphate is followed by the synthesis of the nucleoside monophosphates uridine monophosphate (UMP), cytidine monophosphate (CMP), their deoxy counterparts, and thymidine monophosphate (TMP).
2. The pyrimidine catabolic pathway originates from CMP, UMP, and TMP, and after deamination of cytidine into uridine, it yields β-alanine and β-aminoisobutyrate, which are converted into intermediates of the citric acid cycle.
3. The pyrimidine salvage pathway, which is composed of kinases, converts the pyrimidine nucleosides, cytidine, uridine, and thymidine (which are provided by food intake or the catabolic pathway) into the corresponding nucleotides CMP, UMP, and TMP.
Inborn errors of pyrimidine metabolism comprise errors of pyrimidine nucleotide synthesis, pyrimidine catabolism, and pyrimidine salvage. They cause mostly neurological symptoms, including developmental disability, and muscle wasting.
DISORDERS OF DE NOVO PYRIMIDINE SYNTHESIS
 HEREDITARY OROTIC ACIDURIA (UMP SYNTHASE DEFICIENCY)
HEREDITARY OROTIC ACIDURIA (UMP SYNTHASE DEFICIENCY)
Hereditary orotic aciduria, the first inborn error discovered in pyrimidine metabolism,117 is caused by a deficiency of the last two steps of denovo pyrimidine synthesis—orotate phosphoribosyltransferase (OPRT) and orotidine 5′-monophosphate decarboxylase (ODC). Both steps are catalyzed by a single bifunctional polypeptide called uridine monophosphate (UMP) synthase (Fig. 168-3). Hereditary orotic aciduria is exceedingly rare, with about 20 cases published over nearly five decades. Early recognition of the disorder is mandatory, since it can be easily treated with apparently very good results.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


