Disorders of Platelets
Cindy E. Neunert and Donald L. Yee
PLATELET PRODUCTION, STRUCTURE, AND FUNCTION
Platelets arise from megakaryocytic precursors in the bone marrow. Maturation and production is regulated by several general cytokines and the more specific platelet growth factor, thrombopoietin. Platelet production occurs through the specialized process of endomitosis, by which DNA replicates without cell division, resulting in the polyploid nuclei characteristic of megakaryocytes.1 Proplatelet extensions form from megakaryocytic cytoplasm; once granule and cytoplasmic organization is complete, platelets are released from the ends of the proplatelet structures. After leaving the marrow, approximately one third of the platelet mass is sequestered in the spleen, while the other two thirds circulate with a life span of 7 to 10 days. Thrombopoiesis is balanced by platelet senescence and consumption to maintain a normal blood platelet count (150,000– 400,000/mm3) via the plasma thrombopoietin level. Platelets have an average diameter of 2.0 to 5.0 mm and typical mean platelet volumes of 6 to 10 femtoliters.1 Their external surface consists of a lipid bilayer containing a variety of structural glycoproteins. The anuclear cytoplasm contains dense granules, which store calcium, serotonin, adenosine diphosphate (ADP), and adenosine triphosphate (ATP), and the more numerous alpha granules, which contain other biologically active proteins including fibrinogen, von Willebrand factor, factor V, and other adhesive molecules.
Circulating platelets fulfill many critical hemostatic functions, including adhesion to sites of vascular injury, amplification of the platelet response, secretion of mediators of hemostasis, and aggregation via fibrinogen binding. After vascular injury, these processes lead to formation of a platelet plug, which constitutes primary hemostasis. Platelets also play a central role in activation of the coagulation cascade, or secondary hemostasis. 
Platelet disorders include quantitative processes, affecting platelet number (eg, thrombocytopenia or thrombocytosis), as well as qualitative phenomena, impacting platelet structure or hemostatic function.
THROMBOCYTOPENIA
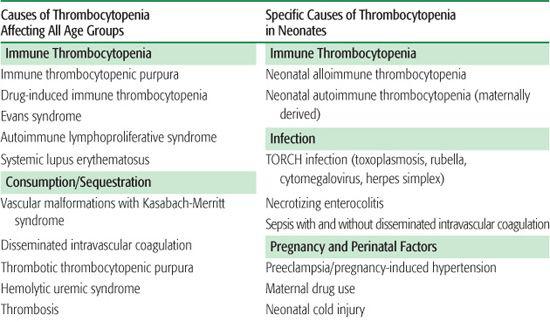
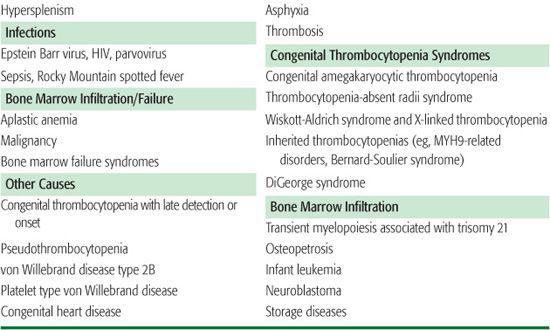
There are three general mechanisms of thrombocytopenia: (1) increased destruction, which may be either immune or nonimmune (consumptive or mechanical); (2) decreased production; or (3) sequestration and pooling. A comprehensive list of causes of thrombocytopenia is outlined in Table 439-1. In some clinical situations, more than one mechanism may be operative. The etiology of thrombocytopenia can often be determined by a detailed history and physical examination, a complete blood count, and a review of the peripheral blood smear. It is also important to ascertain whether the thrombocytopenia is acquired or congenital. A bone marrow examination can often provide diagnostic information if the physical exam and peripheral blood smear are inconclusive. An approach to the child with thrombocytopenia is shown in Figure 439-1.
IMMUNE THROMBOCYTOPENIC PURPURA
 PATHOGENESIS
PATHOGENESIS
Immune thrombocytopenic purpura (ITP) is caused by autoantibodies that interact with platelet surface glycoproteins and lead to increased platelet destruction in the spleen where antibody-coated platelets are removed by mononuclear phagocytes via the Fc receptor.3 The antibodies produced may also interfere with thrombopoiesis. The trigger for this antibody production remains unclear. While immunizations and recent viral illness have been implicated, the mechanism for antibody development is not well understood.
 CLINICAL FEATURES
CLINICAL FEATURES
In children, acute ITP occurs most frequently between the ages of 2 to 10 years. Typical presentation is an otherwise healthy-appearing child with petechiae and purpura; however, oral bleeding, epistaxis, and less commonly, bleeding from the gastrointestinal or genitourinary tracts, may also occur. The child is often previously healthy, with the exception of a recent viral infection or immunizations. A benign history (eg, absence of bone pain and weight loss) and lack of hepatosplenomegaly or lymphadenopathy on physical examination, are important considerations in differentiating ITP from conditions such as leukemia or lymphoma.
 LABORATORY FEATURES
LABORATORY FEATURES
The platelet count varies, but often is less than 20,000/mm3 at presentation. Review of the peripheral blood smear reveals normal leukocyte and red blood cell morphology with normal- to large-sized platelets. Patients with immune thrombocytopenic purpura may be anemic due to significant bleeding; however, alternative diagnoses such as leukemia, aplastic anemia, Evans syndrome, and thrombotic thrombocytopenic purpura should be considered if multiple cytopenias are identified. There is currently no standardized method available to detect the autoantibodies involved in immune thrombocytopenic purpura. Bone marrow examination is not routinely required in the presence of typical laboratory and clinical findings, but when performed, it demonstrates an increased number of megakaryocytes. Levels of the platelet growth factor thrombopoietin are often not increased despite increased peripheral destruction. This observation has led to the development of new therapies with recombinant thrombopoietin analogs.4,5
Table 439-1. Differential Diagnosis of Thrombocytopenia
 CLINICAL COURSE
CLINICAL COURSE
In approximately 75% of children, immune thrombocytopenic purpura (ITP) is a self-limited condition, and complete resolution may occur within 6 to 8 weeks. After remission, the chance of having another episode of ITP is small (1–2%), and following recovery, children are not at increased risk for other blood disorders or cancers. The most feared outcome, intracranial hemorrhage, is fortunately a rare occurrence, with a frequency ranging from 0.3% to 1%.6Children who do not go into spontaneous remission by 6 months are arbitrarily classified as having chronic ITP. There is currently no reliable way to identify those children who will develop chronic ITP, but risk factors include older age at presentation and insidious onset of symptoms.
 TREATMENT
TREATMENT
Treatment of ITP should be individualized based on the patient’s bleeding symptoms and platelet count. Current guidelines7,8 suggest treatment for children with significant hemorrhage and a platelet count less than 10,000 to 20,000/mm3. Treatment to raise the platelet count includes steroids, intravenous immunoglobulin (IVIG) and anti-D immunoglobulin (if the patient is Rh positive).9 Steroids at a dose of 2 to 4 mg/kg enhance vascular stability and decrease autoantibody production and platelet clearance. Side effects of steroids include weight gain, irritability, hypertension, and hyperglycemia. IVIG impairs reticuloendothelial cell clearance of platelets via blockage of Fc receptors. A single dose of 0.8 to 1.0 g/kg commonly leads to an increased platelet count within 24 to 48 hours; side effects include fever and chills during the infusion, headache, and aseptic meningitis. Similar to IVIG, anti-D immunoglobulin is thought to impair Fc receptors, but specifically via binding with antibody-coated red blood cells. Typical dosing is 50 to 75 μg/kg, and like IVIG the response time can be rapid. A decline in hemoglobin concentration by 0.5 to 2.0 g/dL or more can be expected due to induced red blood cell hemolysis, and cases of severe intravascular hemolysis and renal failure with anti-D immunoglobulin have been described.10,11 In the case of severe or life-threatening hemorrhage, prompt management with high-dose methylprednisolone, IVIG, and platelet infusions can help stabilize the patient while emergent splenectomy is considered.
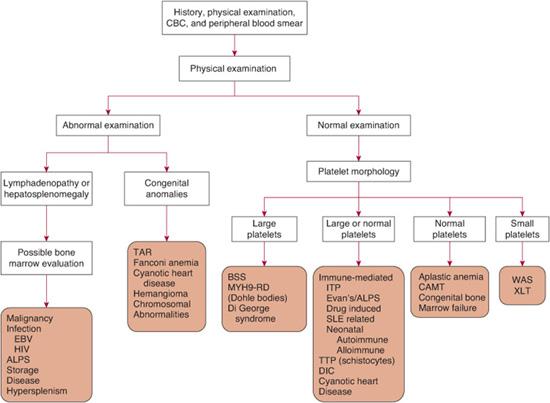
FIGURE 439-1. A diagnostic approach to the patient with thrombocytopenia. While this outlines a general approach, it should be emphasized that exceptions as well as overlap in symptoms and findings can occur, often necessitating additional considerations. ALPS, autoimmune lymphoproliferative disorder; BSS, Bernard-Soulier syndrome; CAMT, congenital amegakaryocytic anemia; CBC, complete blood count; DIC, disseminated intravascular coagulation; EBV, Epstein-Barr virus; HIV, human immunodeficiency virus; ITP, immune thrombocytopenic purpura; MYH9-RD, MYH9-related disorders; SLE, systemic lupus erythematosus; TAR, thrombocytopenia absentradius; TTP, thrombotic thrombocytopenic purpura; WAS, Wiskott-Aldrich syndrome; XLT, X-linked thrombocytopenia.
Management of patients with chronic immune thrombocytopenic purpura (ITP) is based on evaluation of the patient’s platelet count, bleeding severity, and ability to perform daily activities and should occur in consultation with a hematologist. Because approximately one third of patients will have spontaneous remission of their chronic ITP even years after diagnosis,12 management often consists of observation alone, with pharmacologic intervention reserved for severe thrombocytopenia, bleeding episodes, or before anticipated hemostatic challenges (eg, invasive procedures or certain physical activities). However, some children with chronic ITP have more severe thrombocytopenia or significant and frequent bleeding episodes that necessitate more definitive and durable treatment. Options include splenectomy, rituximab, and immunosuppressive agents such as 6-mercaptopurine and cyclosporine. Splenectomy is successful in approximately 70% of children,13 but concerns over the long-term risk of severe infection limits its use. Recently, the monoclonal CD20 antibody rituximab has been reported to elicit a complete response in 30% of patients with chronic ITP.14 Various case reports and small case series have reported success with immunosuppressive agents such as 6-mercaptopurine. New agents that mimic thrombopoietin activity are currently being investigated.4,5
OTHER IMMUNE-MEDIATED THROMBOCYTOPENIAS
 NEONATAL ALLOIMMUNE THROMBOCYTOPENIA
NEONATAL ALLOIMMUNE THROMBOCYTOPENIA
Neonatal alloimmune thrombocytopenia (NAIT) is a common cause of severe, isolated thrombocytopenia in the newborn, with an estimated occurrence of 1 in 1,000 live births.15 Otherwise healthy infants (including firstborns) can present with petechiae, purpura, or mucosal bleeding during the immediate neonatal period.
NAIT results from platelet antigen incompatibility between the mother and fetus (analogous to hemolytic disease of the newborn). Maternal IgG antibodies directed at paternally derived human platelet alloantigens (HPA) cross the placenta and lead to peripheral platelet destruction. The most common antigens involved are HPA-1 a and HPA-5b (∼ 95% of cases).15 These alloanti-bodies bind to the platelet surface glycoprotein GPIIb-IIIa receptors and can inhibit platelet function, which could account for the high rate of severe bleeding complications in this condition as compared to autoimmune thrombocytopenia of the newborn (eTable 439.1  ).
).
NAIT is part of the differential diagnosis of newborns with thrombocytopenia (Table 439-1), along with congenital infection, disseminated intravascular coagulation, congenital heart disease, other congenital disorders, and maternal autoimmune thrombocytopenia. The latter is a key diagnostic consideration and can be evaluated via the maternal platelet count and history. The key differences between the two are shown in eTable 439.1  . NAIT can be diagnosed by specific alloantibody detection using ELISA testing. Specific platelet surface molecule genotyping of the mother and father can identify the antigen incompatibility so that parents may be informed for management of future pregnancies. However, these tests often do not yield timely results to assist with treatment decisions.
. NAIT can be diagnosed by specific alloantibody detection using ELISA testing. Specific platelet surface molecule genotyping of the mother and father can identify the antigen incompatibility so that parents may be informed for management of future pregnancies. However, these tests often do not yield timely results to assist with treatment decisions.
The major source of morbidity and mortality is intracranial hemorrhage (ICH), which occurs with a frequency of 10% to 20%.16 While NAIT is self-limited, with resolution within 2 to 6 weeks, treatment aimed at rapidly raising the platelet count and decreasing the risk of ICH in the immediate postnatal period is critical. Platelet transfusion with maternally derived platelets is preferred; however, lack of availability of maternal platelets should not delay treatment. Random donor platelets have been shown to adequately raise the platelet count to greater than 40,000/mm3 and are suitable for urgent use, but their use may necessitate more frequent transfusions. Intravenous immunoglobulin (IVIG) can increase the platelet count, but because response can take up to 24 to 72 hours, it should be used in conjunction with transfusion therapy. In cases of severe bleeding, when the above measures fail, exchange transfusion has been successful.
Mothers of infants with NAIT must be counseled on the risk during subsequent pregnancies. The risk of ICH in subsequent pregnancies is highest for those women who have had a previous child with ICH from NAIT. 
 MATERNAL AUTOIMMUNE NEONATAL THROMBOCYTOPENIA
MATERNAL AUTOIMMUNE NEONATAL THROMBOCYTOPENIA
Infants born to mothers with a history of immune thrombocytopenic purpura may experience thrombocytopenia at birth due to transfer of maternal antiplatelet IgG across the placenta into the fetal circulation. The degree of maternal thrombocytopenia is unrelated to the infant’s platelet count. The risk of serious bleeding is significantly less than that associated with neonatal alloimmune thrombocytopenia (eg, intracranial hemorrhage frequency of 0.5–1%). In utero assessment of platelet count is not indicated, but a cord blood platelet count should be performed at birth and repeated periodically as platelet counts can decline with a nadir around 4 days of age. The majority of hemorrhagic events occur within 24 to 48 hours after birth, so infants should be monitored closely during this period and consideration given to appropriate imaging studies.
If significant hemorrhage or severe thrombocytopenia (< 20,000/mm3) is encountered, intravenous immunoglobulin is the treatment of choice. Thrombocytopenia typically lasts for 1 to 2 months, reflecting the survival of maternal IgG in the infant. Subsequent births are also at risk.
 THROMBOCYTOPENIA ASSOCIATED WITH OTHER IMMUNE DISORDERS
THROMBOCYTOPENIA ASSOCIATED WITH OTHER IMMUNE DISORDERS
Evans syndrome is characterized by multiple immune cytopenias. Classic Evans syndrome is the combination of immune thrombocytopenic purpura and Coombs-positive hemolytic anemia; however, neutropenia secondary to antineutrophil antibodies may also occur. Typically, Evans syndrome is treated acutely with steroids, which are tapered based on the patient’s hematologic response.
Underlying causes of Evans syndrome include systemic lupus erythematous and autoimmune lymphoproliferative disorder (ALPS). ALPS results from impaired lymphocyte apoptosis leading to lymphoid proliferation and autoimmune dysfunction.
 DRUG-INDUCED THROMBOCYTOPENIA
DRUG-INDUCED THROMBOCYTOPENIA
The most common drugs causing thrombocytopenia are heparin, quinine/quinidine, sulfa derivatives, gold, and valproic acid. Thrombocytopenia results from either binding of drug to the platelet followed by binding of antidrug antibody or by direct binding of a drug and antidrug complex to the platelet surface. Drug withdrawal leads to a prompt rise in the platelet count. Heparin-induced thrombocytopenia (HIT) is relatively uncommon in children.  Thrombocytopenia develops 5 to 14 days after heparin exposure, and venous or arterial thrombosis can occur as a result of platelet activation. Enzyme-linked immunosorbent assay (ELISA) testing can identify the antibody, but the result should be carefully interpreted within the clinical context as false positives can occur. Discontinuation of all heparin and use of an alternative anticoagulant is standard treatment for HIT and should be initiated when testing is performed.
Thrombocytopenia develops 5 to 14 days after heparin exposure, and venous or arterial thrombosis can occur as a result of platelet activation. Enzyme-linked immunosorbent assay (ELISA) testing can identify the antibody, but the result should be carefully interpreted within the clinical context as false positives can occur. Discontinuation of all heparin and use of an alternative anticoagulant is standard treatment for HIT and should be initiated when testing is performed.
 PSEUDOTHROMBOCYTOPENIA
PSEUDOTHROMBOCYTOPENIA
Pseudothrombocytopenia is a cause of spurious thrombocytopenia in an otherwise healthy individual with no clinical findings consistent with thrombocytopenia. It results from in vitro ethylene diamine tetracetic acid (EDTA)-dependent platelet aggregation and clumping.17 The abnormalities can be confirmed by repeating the platelet count using an alternative anticoagulant such as citrate.
INCREASED PLATELET DESTRUCTION/CONSUMPTION
Consumptive coagulopathy from infection or other causes (eg, trauma, malignancy) can lead to thrombocytopenia. Clues suggestive of disseminated intravascular coagulation include elevated prothrombin time/activated partial thromboplastin time and D-dimers, low fibrinogen levels, microangiopathic hemolysis, and the presence of an underlying condition known to be associated with disseminated intravascular coagulation. Treatment of the underlying condition is required as well as blood product support to minimize bleeding risk.
Platelet destruction can also occur within vascular lesions (typically kaposiform hemangioendotheliomas or tufted angiomas) of the skin, liver, or spleen, and some patients may even have evidence of a consumptive coagulopathy (Kasabach-Merritt syndrome). Management of the lesion depends on its size, obstruction of major vessels or organs, and severity of the coagulopathy.
 THROMBOTIC THROMBOCYTOPENIC PURPURA
THROMBOTIC THROMBOCYTOPENIC PURPURA
Thrombotic thrombocytopenia purpura results from inadequate cleavage of von Willebrand factor (vWF) multimers due to either congenital absence of or acquired antibody to the specific vWF–cleaving metalloprotease, ADAMTS13. The resulting large vWF multimers promote formation of platelet microthrombi in the microvasculature. The classic diagnostic pentad for thrombotic thrombocytopenic purpura (TTP) comprises microangiopathic hemolytic anemia, thrombocytopenia, neurologic findings, renal manifestations, and fever; however, not all features need be present. Laboratory findings include schistocytes on the peripheral smear, an elevated lactic dehydrogenase (LDH) and reticulocyte count, and thrombocytopenia. Testing for metalloprotease levels and the presence of antibodies is available, but it should not delay diagnosis and treatment because mortality without treatment approaches 80%. Recommended therapy for congenital TTP involves plasma infusions, whereas acquired TTP is treated urgently with plasma exchange.18 Hemolytic uremic syndrome is thought to exist on a continuum with TTP and thus may feature thrombocytopenia, but it exhibits more extensive renal involvement and does not require plasma exchange.
DECREASED PLATELET PRODUCTION
 CONGENITAL THROMBOCYTOPENIAS
CONGENITAL THROMBOCYTOPENIAS
Congenital thrombocytopenias represent a group of disorders in which thrombocytopenia can be present at birth and varies in severity.17,19 As outlined below these conditions are often associated with other physical findings or disorders.
MYH9-Related Disorders
The MYH9-related disorders represent a group of autosomal dominant macrothrombocytopenias characterized by mutations in the MYH9 gene, which encodes the non-muscle myosin heavy chain II.20 Pathognomonic Döhle-like bodies (light blue inclusions in neutrophils) can be seen on light microscopy. Patients should be screened for additional clinical manifestations such as sensorineural hearing loss, cataracts, and renal failure, as these features may be subtle. Mutation analysis of the MHY9 gene can confirm the diagnosis. Platelet transfusions may be given for significant bleeding events and as prophylaxis for surgery. Other causes of macrothrombocytopenia are outlined in Figure 439-1.
Wiskott-Aldrich Syndrome and X-Linked Thrombocytopenia
Wiskott-Aldrich syndrome (WAS) and the less severe X-linked thrombocytopenia (XLT) are X-linked disorders associated with micro-thrombocytopenia caused by mutations in the WAS gene located at Xp11.22-11.23.21 Bleeding manifestations during infancy may bring patients to medical attention and continue throughout life, with 30% of patients encountering at least 1 severe bleeding episode.22 Patients with WAS, unlike those with XLT, also have impaired cellular and humoral immunity and are at increased risk for bacterial, viral, and fungal infections and early development of lymphoreticular malignancies. WAS and XLT should be suspected in any male patient with thrombocytopenia and small platelets on the peripheral blood smear. Consultation with an immunologist should be undertaken in order to screen for both cellular and humoral immune dysfunction. 
Bleeding symptoms associated with WAS and X-linked thrombocytopenia can be managed with platelet transfusions, and splenectomy in severe cases. However, given the risk of life-threatening infections, early stem cell transplantation is considered standard of care for any patient with a matched sibling or an excellent unrelated HLA-matched donor. Recent attention has been given to the development of gene therapy for patients with WAS, and current animal and human cell models show promise.
Thrombocytopenia-Absent Radii Syndrome
Thrombocytopenia-absent radii syndrome (TAR) is characterized by severe thrombocytopenia (< 50,000/mm3) at birth and bilateral absence of radii, but presence of thumbs.25 The patho-physiology and genetic basis for TAR remain obscure. Additional features include cardiac and facial anomalies, mental retardation, and renal anomalies. There is a high frequency of cow’s milk allergy associated with TAR, and it is recommended that children avoid cow’s milk formula for the first year of life.25 Although more severe thrombocytopenia can be triggered by infection and diet, the platelet counts tend to improve with age. Any child with bilateral absent radii should be evaluated with a platelet count. Due to fluctuating platelet counts, the diagnosis should not be excluded on the basis of a single normal value. Bone marrow evaluation will demonstrate decreased to absent megakaryocytes. Platelet transfusions for bleeding episodes are the mainstay of treatment.
Congenital Amegakaryocytic Thrombocytopenia
Congenital amegakaryocytic thrombocytopenia (CAMT) is a rare autosomal recessive disorder of platelet production that presents with severe thrombocytopenia in the newborn period. Unlike patients with thrombocytopenia absent radii syndrome, skeletal findings are absent. Thrombocytopenia results from a mutation in the thrombopoietin receptor, which renders it unresponsive to thrombopoietin.26 Platelets are normal in size. Patients suspected to have CAMT should undergo a bone marrow evaluation. Unlike patients with thrombocytopenia absent radii syndrome, the thrombocytopenia of CAMT does not improve with age, and many patients develop aplastic anemia or leukemia within the first 5 years of life.27 Initial management is aimed at the prevention and control of hemorrhage via platelet transfusions. Early stem cell transplantation should be considered, particularly if there is a matched sibling donor, because of the high rate of malignant transformation.
 BONE MARROW FAILURE AND INFILTRATION
BONE MARROW FAILURE AND INFILTRATION
Bone marrow failure and infiltration should be considered in any patient with multiple cytopenias. The differential diagnosis is extensive and includes acquired aplastic anemia, Fanconi anemia, parvovirus (or other viral suppression), leukemia, and osteopetrosis. Each of these entities is discussed more extensively in their respective chapters.
 SPLENIC SEQUESTRATION AND POOLING
SPLENIC SEQUESTRATION AND POOLING
Increased platelet sequestration occurs in the setting of hypersplenism and is often accompanied by other cytopenias. Causes of hypersplenism include portal hypertension, portal vein thrombosis, storage diseases, hematological disorders such as sickle cell anemia, and infiltrative diseases of the liver and spleen (see below). Virtually all patients have marked splenomegaly.
THROMBOCYTOSIS
Thrombocytosis (defined as a platelet count exceeding 500 × 103/mm3) in children usually occurs secondary to another condition (Table 439-2). Such reactive thrombocytosis has been reported to occur in up to 15% of hospitalized children.28 A careful history and physical examination can frequently help identify an underlying diagnosis; acute phase reactants may also be increased. As a secondary process, reactive thrombocytosis tends to be transient,29 with normalization of the platelet count coinciding with resolution of the primary condition. High platelet counts (even surpassing 1000 × 103/mm3) can be encountered under these circumstances and are usually not thought to be associated with increased risk of thrombosis, unless they occur following splenectomy in conjunction with other prothrombotic risk factors. Treatment should be directed at the underlying disorder,30 but thromboprophylaxis or other intervention may warrant consideration if the patient has coexisting risk factors such as those mentioned above.
A careful history and physical examination can frequently help identify an underlying diagnosis; acute phase reactants may also be increased. As a secondary process, reactive thrombocytosis tends to be transient,29 with normalization of the platelet count coinciding with resolution of the primary condition. High platelet counts (even surpassing 1000 × 103/mm3) can be encountered under these circumstances and are usually not thought to be associated with increased risk of thrombosis, unless they occur following splenectomy in conjunction with other prothrombotic risk factors. Treatment should be directed at the underlying disorder,30 but thromboprophylaxis or other intervention may warrant consideration if the patient has coexisting risk factors such as those mentioned above.
Table 439-2. Causes of Thrombocytosis



