Disorders of Metabolism of Neurotransmitter Amino Acids
Jaak Jaeken
 INTRODUCTION
INTRODUCTION
This chapter deals with defects in the catabolism of gamma-aminobutyric acid (GABA), with defects in receptors of neurotransmitters (GABA and glycine) and in monoamine metabolism. (Glutamine synthase deficiency is discussed in Chapter 145, pyridoxine responsive disorders in Chapter 149, and glycine/serine disorders in Chapter 139.)
Two genetic defects are known in GABA catabolism (Fig. 141-1): the extremely rare, severe, and untreatable GABA transaminase deficiency, and the much more frequent and, to some extent, treatable succinic semialdehyde dehydrogenase (SSADH) deficiency.
Hyperekplexia is a dominantly inherited defect in subunits of the glycine receptor (α1 and β) and in the receptor-associated protein gephyrin. This disease is characterized by excessive startle responses, which are treatable with clonazepam. Mutant GABAA receptor γ-2 subunit causes absence epilepsy and febrile seizures.
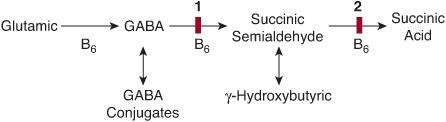
FIGURE 141-1. Brain metabolism of GABA. B6: pyridoxal phosphate coenzyme; 1: GABA transaminase; 2: succinic semialdehyde dehydrogenase. Enzyme defects are indicated by solid red bars.
Nine defects have been reported in the metabolism of monoamines (Figs. 141-2 and 141-3): five in the synthesis of the cofactor tetrahydrobiopterin, tyrosine-hydroxylase (TH) deficiency, aromatic amino acid decarboxylase (AADC) deficiency, dopamine β-hydroxylase (DBH) deficiency, and monoamine oxidase A (MAO-A) deficiency. All are associated with neurological symptoms except DBH deficiency (orthostatic hypotension). With the exception of MAO-A deficiency, they are more or less efficiently treatable.
GABA CATABOLISM DEFECTS
 GABA TRANSAMINASE DEFICIENCY
GABA TRANSAMINASE DEFICIENCY
Clinical Presentation
GABA transaminase deficiency was reported in 1984 in a brother and sister from a Flemish family. No other patients have been reported. The siblings showed feeding difficulties from birth, often necessitating gavage feeding; profound axial hypotonia; and generalized convulsions. Hyperreflexia was present during the first 6 to 8 months. Psychomotor development was nearly absent. Growth acceleration was present from birth, due to growth hormone hypersecretion. Postmortem examination of the brain showed spongiform leukodystrophy.
Metabolic Derangement
The CSF and plasma concentrations of GABA, GABA conjugates, and β-alanine were increased due to a decrease of GABA transaminase activity. β-alanine is an alternative substrate for GABA transaminase, which explains its increase in this disease.
Genetics
GABA transaminase deficiency (OMIM number: 137150) is an autosomal recessive disease. The patients were compound heterozygotes for two missense mutations of GABAT on chromosome 16p13.3.
Diagnostic Tests
The diagnosis requires amino acid analysis of the CSF. Since free GABA CSF levels are low, sensitive techniques must be used, such as ion-exchange chromatography with fluorescence detection or a stable isotope dilution technique. Enzymatic confirmation is obtained using lymphocytes, lymphoblasts, or liver. Prenatal diagnosis is possible by enzymatic analysis of chorionic villus tissue, not of amniocytes.
Treatment
No efficient treatment is available. The siblings died at the ages of 1, 2, and 7 years.1
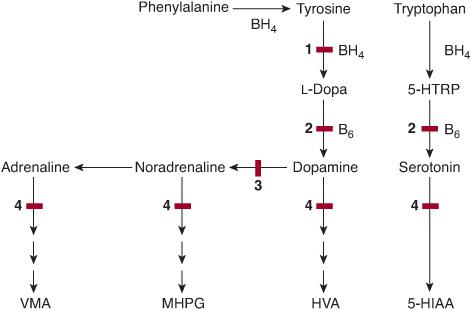
FIGURE 141-2. Monoamine metabolism. 5-HTRP: 5-hydroxytryptophan; BH4: tetrahydrobiopterin; B6: pyridoxal phosphate coenzyme; VMA: vanillylmandelic acid; MHPG: 3-methoxy-4-hydroxyphenylglycol; HVA: homo-vanillic acid; 5-HIAA: 5-hydroxyindole acetic acid; 1: tyrosine hydroxylase; 2: aromatic L-amino acid decarboxylase; 3: dopamine beta hydroxylase; 4: monoamine oxidase. Enzyme defects are indicated by solid red bars.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


