Disorders of Lipid and Lipoprotein Metabolism
Peter O. Kwiterovich Jr.
Disorders of lipid and lipoprotein metabolism are characterized by dyslipidemia, which is defined as either elevated or low levels of one or more of the major lipoprotein classes: chylomicrons, very-low-density lipoproteins (VLDL), low-density lipoproteins (LDL), and high-density lipoproteins (HDL). Dyslipidemia can result from the expression of a mutation in a single gene that plays a paramount role in lipoprotein metabolism. More often, dyslipidemia reflects the influence of multiple genes. Environmental influences such as excessive dietary intake of fat and calories and limited physical activity, particularly when associated with overweight or obesity, can also contribute significantly to dyslipidemia.  The major clinical complication of dyslipidemia is a predilection to atherosclerosis starting early in life and leading to cardiovascular disease (CVD) in adulthood. At the extremes of dyslipidemia, where inherited disorders of lipid and lipoprotein metabolism are more likely to occur, premature CVD is more frequent and can be accompanied by deposition of lipid in various tissues. Children with profound hypertriglyceridemia are at high risk of pancreatitis.
The major clinical complication of dyslipidemia is a predilection to atherosclerosis starting early in life and leading to cardiovascular disease (CVD) in adulthood. At the extremes of dyslipidemia, where inherited disorders of lipid and lipoprotein metabolism are more likely to occur, premature CVD is more frequent and can be accompanied by deposition of lipid in various tissues. Children with profound hypertriglyceridemia are at high risk of pancreatitis.
 BACKGROUND
BACKGROUND
A number of clinical, epidemiological, metabolic, genetic, and randomized clinical trials strongly support the tenet that the origins of atherosclerosis and CVD risk factors begin in childhood and adolescence and that treatment should begin early in life.1
Several longitudinal pathological studies from the general population have found that early atherosclerotic lesions of fatty streaks and fibrous plaques in children, adolescents, and young adults who died from accidental causes are significantly related to higher antecedent levels of total cholesterol (TC) and LDL-C (LDL-C); to lower levels of HDL-C (HDL-C); and to other CVD risk factors such as obesity, higher blood pressure, and cigarette smoking.1 These risk factors’ effects on coronary lesion severity are multiplicative rather than additive.
Four major prospective population studies from Muscatine, Bogalusa, the Coronary Artery Risk Development in Young Adults (CARDIA) and the Special Turku Coronary Risk Factor Intervention Project (STRIP) showed that CVD risk factors in children and adolescents, particularly LDL-C and obesity, predicted clinical manifestations of atherosclerosis in young adults. 
Studies have also been performed in high-risk youth; these individuals were selected because one parent had CVD or because they have inherited a known metabolic disorder of lipoprotein metabolism that produces premature CVD. Half of the young progeny of men who had CVD before age 50 had one of seven dyslipidemic profiles. 
Examples of inherited lipoprotein disorders that often present in youth at high risk of future CVD include familial hypercholesterolemia (FH; which is caused by a defect in the LDL receptor, LDLR); familial combined hyperlipidemia (FCHL); and its metabolic cousin hyperapoB, the prototypes for hepatic overproduction of VLDL. 
 LIPOPROTEIN CLASSIFICATION AND PROPERTIES
LIPOPROTEIN CLASSIFICATION AND PROPERTIES
Plasma lipoproteins are spherical particles consisting of a core of nonpolar lipids—TG and cholesteryl ester—surrounded by a surface coating consisting of proteins (apolipoproteins) and more polar lipids, phospholipids, and unesterified (free) cholesterol. Plasma lipoproteins are classified by their density and electrophoretic mobility into four major groups: chylomicrons, VLDL, LDL, and HDL (Table 166-1). After electrophoresis, chylomicrons remain at the origin, and VLDL, LDL, and HDL migrate in the same positions as pre-β-, β-, and α-globulins, respectively. The hydrated density of the lipoproteins is related to their chemical composition and the relative content of lipid and apolipoprotein. Chylomicrons are 99% lipid, most of it being TG (Table 166-1). After plasma has stood overnight, these large particles (80–500 nm) will rise to the top, where they appear as a creamy layer. VLDL is about 90% lipid, the majority of it being TG, with lesser amounts of cholesterol. When present in plasma in increased amounts, VLDL are large enough (30–80 nm) to create a cloudy or turbid appearance to plasma. LDL are the major carriers of cholesterol in plasma, and about 50% of their weight is cholesteryl ester and cholesterol. HDL comprise about equal amounts of apolipoprotein and lipid, principally phospholipids and cholesterol.
 APOLIPOPROTEINS
APOLIPOPROTEINS
Lipoproteins are associated with several apolipoproteins (Table 166-2). Nomenclature for the apolipoproteins follows an alphabetical scheme.  The nucleotide sequences of cDNA for the apolipoproteins have been determined.
The nucleotide sequences of cDNA for the apolipoproteins have been determined.
 ORIGIN AND FATE OF PLASMA LIPIDS AND LIPOPROTEINS
ORIGIN AND FATE OF PLASMA LIPIDS AND LIPOPROTEINS
The transport of plasma lipids by lipoproteins may be divided into exogenous (dietary) and endogenous systems (Fig. 166-1).
Exogenous Lipid Transport
Most dietary lipid is in the form of neutral fat or TG (75–150 g/d). The amount of cholesterol in the diet is usually about 300 mg/day but varies from 100 to 600 mg/day. In addition to dietary cholesterol, about 1100 mg of biliary cholesterol is secreted each day from liver into the intestine (Fig. 166-1). In the small intestine, lipids are emulsified by bile salts and hydrolyzed by pancreatic lipases. The bile acids are then reabsorbed by the intestinal bile acid transporter (IBAT) for return to the liver through the enterohepatic pathway (Fig. 166-1). TG is broken down into fatty acids and 2-monoglycerides; cholesteryl ester is hydrolyzed into fatty acids and unesterified cholesterol. These components are then absorbed by the intestinal cells. The absorption of cholesterol occurs in the jejunum, through the high-affinity uptake of dietary and biliary cholesterol by the Niemann-Pick C-1L-1 (NP-C-1L-1) protein (Fig. 166-1). Normally, about half the dietary and biliary cholesterol is absorbed daily. Excessive cholesterol absorption is prevented by the ABCG5/ABCG8 transporters, which act together to pump excess cholesterol and plant sterols from the intestine back into the lumen for excretion into the stool (Fig. 166-1).
Table 166-1. Classification and Properties of the Major Human Plasma Lipoproteins
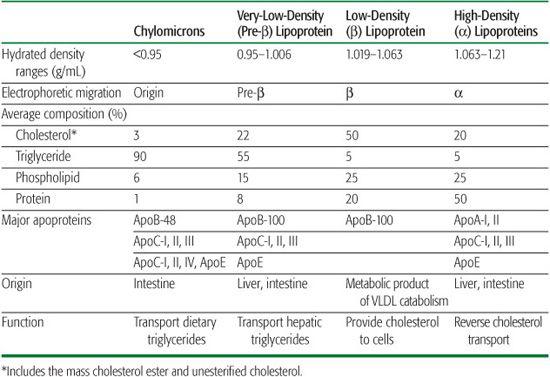
Table 166-2. Classification and Properties of Major Human Plasma Apolipoproteins
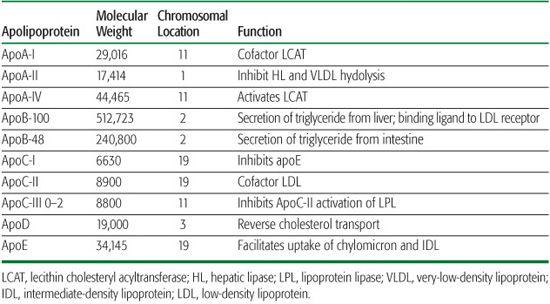
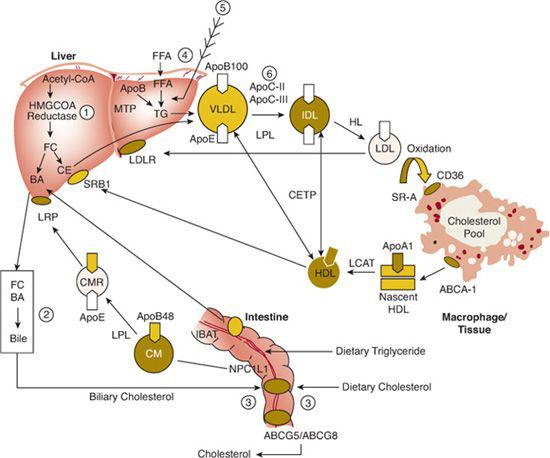
FIGURE 166-1. Overview of lipoprotein metabolism. Three major pathways of plasma lipoprotein metabolism are shown: transport of dietary (exogenous) fat (left); transport of hepatic (endogenous) fat (center); reverse cholesterol transport (bottom). A detailed description appears in the text. The sites of action of the six major lipid-altering drugs on exogenous and endogenous pathways of lipoprotein metabolism are (1) inhibition of hydroxymethylglutaryl (HMG) CoA reductase by statins; (2) binding of bile acids by sequestrants, interfering with their reabsorption by the ileal bile acid transporter (IBAT); (3) binding of a cholesterol absorption inhibitor to the Niemann-Pick C1L1, decreasing the absorption of dietary and biliary cholesterol; (4) decreased mobilization of free fatty acids (FFA) by nicotinic acid, leading to decreased uptake of FFA by liver and reduced VLDL, IDL, and LDL production; (5) inhibition of TG synthesis by omega-3 fatty acids; (6) upregulation of lipoprotein lipase (LPL) and decreased production of apoC-III, an inhibitor of LPL, by a fibric acid derivative, leading to decreased VLDL-TG. The hepatic cholesterol pool is decreased by the agents at steps 1, 2, and 3, each leading to an upregulation of the LDLR.
In intestinal cells, monoglycerides are reesterified into TG, and cholesterol is esterified by acyl cholesterol acyltransferase (ACAT). Both lipids are packaged into chylomicrons, along with apolipoproteins apoA-I, apoA-II, apoA-IV, and apoB-48. Chylomicrons are secreted into the thoracic duct; from there, they enter the peripheral circulation, where they acquire apoC-II and apoE from HDL. Chylomicrons are too large to cross the endothelial barrier, and apoC-II, a cofactor for lipoprotein lipase (LPL), facilitates the hydrolysis of TG near the endothelial lining of blood vessels. The fatty acids that are released are taken up by muscle cells for energy utilization or by adipose cells for re-esterification into TG. As a result, a chylomicron remnant is produced that is enriched in cholesteryl ester and apoE. This remnant is rapidly taken up by the liver by receptor-mediated endocytosis of remnants through the interaction of apoE with the chylomicron remnant receptor (LRP), or the LDLR on the surface of parenchymal cells (Fig. 166-1).
The uptake of dietary and biliary cholesterol is part of a process that regulates the pool of hepatic cholesterol by downregulating the LDLR and by inhibiting the rate-limiting enzyme of cholesterol biosynthesis, hydroxymethylglutaryl (HMG)-CoA reductase (see also below).
Endogenous Lipid Transport
In the fasting state, most TG in plasma is carried by VLDL. TG is synthesized in the liver, packaged into VLDL with other lipids and apolipoproteins (Table 166-1)—primarily apoB-100, apoE, apoC-I, apoC-II, and apoC-III—and secreted into plasma. VLDL TG is subsequently hydrolyzed by LPL and its cofactor apoC-II to produce VLDL remnants and then intermediate-density lipoproteins (IDL; d, 1.006–1.019 g/mL). TG can be transferred from VLDL and IDL to HDL and LDL in exchange for cholesteryl ester by the cholesterol ester transfer protein (CETP) (Fig. 166-1). Compared with VLDL, IDL are relatively enriched in cholesteryl ester and depleted in TG. Some IDL are taken up directly by the liver, but others are hydrolyzed by hepatic lipase (HL) to produce LDL, the final end product of VLDL metabolism (Fig. 166-1).
The apoB-100 component of the cholesteryl ester-rich LDL are recognized and bound by the high-affinity LDLR either in the liver or in extrahepatic cells (Fig. 166-1). The bound LDL are internalized by absorptive endocytosis. In lysosomes, apolipoprotein B-100 is broken down into amino acids, cholesteryl esters are hydrolyzed, and unesterified cholesterol are released. Cholesterol mediates the proteolytic release of a transcription factor, the sterol regulatory element binding protein (SREBP), from the endoplasmic reticulum (ER).2 This effect occurs through the SREBP cleavage-activating protein (SCAP) that is both a sensor of sterols and an escort of SREBP. For example, when hepatocytes are depleted of cholesterol, SCAP transports SREBP from the ER to the Golgi, where two proteases—site-1 protease and site-2 protease—act in sequence to release the NH2-terminal of SREBP from the membrane.2 The NH2-terminal of SREBP containing the bHLH-zip domain of SREBP enters the nucleus and binds to a sterol response element (SRE) in the promoter area of the LDLR and HMG-CoA reductase genes, increasing their transcription. As the cholesterol content of the hepatocyte increases, the SREBP/SCAP complex is not incorporated into the ER, SREBP cannot reach the Golgi, the NH2-terminal domain of SREBP cannot be released from the membrane for transport into the nucleus, and the transcription of the LDLR and HMG-CoA reductase genes decreases.2
This pathway has important clinical implications. For example, excess dietary and biliary cholesterol leads to the downregulation of the LDLR and HMG-CoA reductase and an increase in LDL-C. Dietary saturated fat content has an even more profound effect on LDL-C than dietary cholesterol. When cholesterol is reesterified by ACAT, SCAP senses a decrease in hepatic cholesterol, leading to the upregulation of the LDLR and HMG-CoA reductase genes by SREBP. However, the preferred substrate for ACAT is oleic acid. Thus, excess saturated fatty acids decrease ACAT activity and thereby increase unesterified cholesterol, which inhibits the proteolysis and release of SREBP and thereby downregulates the LDLR and HMG-CoA reductase genes, followed by an increase in LDL-C. Decreasing dietary cholesterol and saturated fatty acids or decreasing the hepatic cholesterol content with drugs, such as cholesterol absorption inhibitors and the bile acid sequestrants (Fig. 166-1), leads to an upregulation of LDLR and HMG-CoA reductase genes and lower LDL-C. Inhibitors of HMG-CoA reductase (the statins) also reduce the liver’s cholesterol content, leading to an upregulation of LDLR but without the concomitant increase in HMG-CoA reductase activity (Fig. 166-1).
When plasma LDL-C exceeds 100 mg/dL, the capacity to process LDL through the LDLR pathway is exceeded. Increased numbers of LDL particles cross the endothelial barrier; LDL are trapped in the vascular wall by proteoglycans and are then modified by either oxidation or glycation. Such modified LDL binds to the scavenger receptors CD36 and SRA (Fig. 166-1) and enter cells such as macrophages by a low-affinity, LDL-receptor-independent mechanism. This alternate pathway is not subject to feedback inhibition of LDLR synthesis by LDL-derived cholesterol. Thus, LDL continues to be taken up in an unregulated fashion, leading to excess deposition of cholesterol and cholesteryl ester in macrophages (Fig. 166-1). Dyslipidemias that favor an increased uptake of LDL through the scavenger pathway promote the production of foam cells and the associated atherosclerosis and xanthomas.
Reverse Cholesterol Transport
HDL are synthesized as nascent particles primarily in the liver but also in the intestine. After entering plasma, HDL participates in two important reactions. In the process of lipolysis, apoA-I is transferred from chylomicrons to HDL, and apoC-II and apoE on HDL are transferred to the TG-rich lipoproteins. ApoA-I is a cofactor for the enzyme lecithin cholesterol acyltransferase (LCAT; see Tables 166-1 and 166-2). Unesterified cholesterol is removed from peripheral cells through the ATP-binding cassette (ABC) protein ABCA1 and esterified through the action of LCAT and apoA-I (Fig. 166-1). These cholesteryl esters are then transferred from HDL to the apoB-containing lipoproteins by CETP, from which they are taken up by LDLR and LRP (Fig. 166-1). Cholesteryl ester may also be delivered directly to the liver through an HDL receptor (SRB1). These reactions reflect a process called reverse cholesterol transport and may explain the protective effect that HDL and apoA-I have against the development of atherosclerosis. Conversely, factors that impede this process appear to promote atherosclerosis.
SCREENING FOR DYSLIPIDEMIA IN YOUTH
Two major approaches have been considered to detect dyslipidemia in youth, namely screening in the general population or in a selected population. 
Traditionally, screening for dyslipidemias in high-risk children was recommended, because they have multiple CVD risk factors or a family history of premature CVD and/or hypercholesterolemia. LDL-C has been the main focus of diagnosis and treatment. Less attention has been paid to HDL-C and TG. Now, with obesity and the metabolic syndrome evident in our youth,1,4 the focus of screening will likely include other factors such as obesity, low HDL-C, non-HDL-C (TC minus HDL-C), elevated TG, elevated apoB (reflecting increased small dense LDL-P), glucose intolerance and insulin resistance, and higher blood pressure levels. Both the current and evolving concepts in screening for dyslipidemia in youth will now be discussed.
 WHO TO SCREEN
WHO TO SCREEN
Selective Screening
In 1992, the National Cholesterol Education Program (NCEP) Expert Panel on Blood Cholesterol Levels in Children and Adolescents5 recommended that selective, not general, screening be performed. We have expanded some of these recommendations (in italics) in the following NCEP guidelines for screening:
1. A lipoprotein profile in youth whose parents and/or grandparents required coronary artery bypass surgery or balloon angioplasty prior to age 55 years
2. A lipoprotein profile in those with a family history of myocardial infarction, angina pectoris, peripheral or cerebral vascular disease, or sudden death prior to age 55
3. A TC in those whose parents have high TC levels (>240 mg/dl). This recommendation might be usefully expanded to a lipoprotein profile in offspring of parents who have any dyslipidemia involving elevated LDL-C, non-HDL-C, apoB, TG, or low HDL-C.
4. A lipoprotein profile if the parental/grandparental family history is not known, and the patient has two or more other risk factors for CAD, including obesity (BMI > 30), hypertension, cigarette smoking, low HDL-C, physical inactivity, and diabetes mellitus. A new recommendation for a specific category is proposed here: A lipoprotein profile if either obesity (BMI > 95th percentile) or overweight (BMI 85 to 94th percentile) is detected, regardless of the presence of other non-lipid CVD risk factors.
Universal Screening
Universal lipid screening of all children is controversial.3,5
Ideally, each child and adolescent should have an assessment of their plasma lipids and lipoproteins. While there are practical problems (see below), and no longitudinal studies are available to show that treatment starting in childhood decreases adult CVD,3 one might argue that universal screening seems all the more urgent, given the epidemic of obesity and metabolic syndrome in American youth. 
 WHAT TO MEASURE
WHAT TO MEASURE
For selective screening, a lipoprotein profile after an overnight fast is measured for youth who have a positive family history of premature CVD or dyslipidemia, obesity, multiple CVD risk factors, and for those suspected of having secondary dyslipidemia. Such a profile includes TC, TG, LDL-C, HDL-C, and non-HDL-C. Levels of lipoproteins are typically measured and expressed in terms of their cholesterol content. LDL-C is calculated from the Friedewald equation: LDL-C = TC – HDL-C – (TG/5). Total TG in the fasting state divided by 5 is used to estimate the levels of VLDL-C. If the TG is >400 mg/dl, this formula cannot be used and a direct LDL-C may be measured. If the patient is nonfasting, TC HDL-C and non-HDL-C levels can be measured.
ApoB and apoA-I might also be determined, using well-standardized immunochemical methods.8,9 Such measurements might provide additional useful information, particularly in youth whose parents have premature CAD.1 ApoB provides an assessment of the total number of apoB-containing lipoprotein particles.9
ApoB provides an assessment of the total number of apoB-containing lipoprotein particles.9
Non-HDL-C
Non-HDL-C is determined by subtracting HDL-C from TC and can be measured in plasma from nonfasting patients. Non-HDL-C reflects the amount of cholesterol carried by the “atherogenic” apoB-containing lipoproteins (VLDL, IDL, LDL, and Lp [a]). In adults, non-HDL appears to be a better independent predictor of CVD than LDL-C.9 In children, non-HDL-C is at least as good a predictor as LDL of future dyslipidemia in adulthood.1
Summary For universal screening, the simplest approach is measuring TC, HDL-C, and non-HDL in nonfasting patients. However, treatment algorithms in pediatrics are usually focused on fasting LDL-C. HyperTG is usually assessed as part of the dyslipidemic triad and is often elevated in obesity and the metabolic syndrome.1,4 Thus, in an ideal screening program, TC, TG, LDL-C, HDL-C, and non-HDL-C would be assessed by performing a lipoprotein profile in the fasting state.
 WHEN TO SAMPLE FOR DYSLIPIDEMIA
WHEN TO SAMPLE FOR DYSLIPIDEMIA 
Therefore, screening for dyslipidemia is not generally recommended before 2 years of age. After 2 years of age, the levels of the lipids and lipoproteins become quite constant up to adolescence.5
Ten years of age has been proposed as a good time to obtain a lipoprotein profile.6 Children this age are able to fast easier, the values are predictive of future adult lipoprotein profiles, and adolescence has not yet set in. Since TC and LDL-C may fall 10% to 20% (or more) during adolescence,1 it is preferable to screen children at risk for familial dyslipidemias before adolescence, between ages 2 and 10. Even in FH heterozygotes, there is a significant fall in the 1:1 ratio of affected to normal in adolescence.1 If sampling occurs during adolescence and the results are abnormal, then they are likely to be even higher after adolescence. If the results during adolescence are normal, then sampling will need to be repeated toward the end of adolescence (for girls, age 16 and for boys, age 18). 
 DEFINITION OF DYSLIPIDEMIA
DEFINITION OF DYSLIPIDEMIA
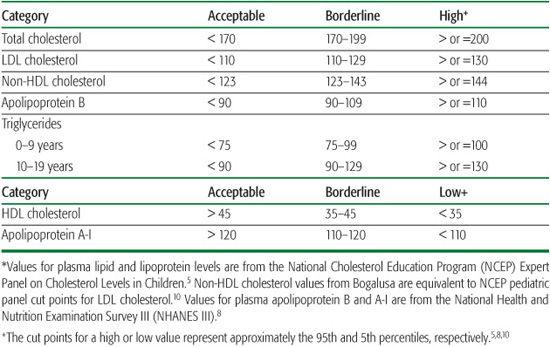
Cut points to define elevated TC, LDL-C, apoB, non-HDL-C, and TG, and low HDL-C and apoA-I in children and adolescents are found in Table 166-3. Dyslipidemia is present if one or more of these lipid, lipoprotein, or apolipoprotein factors are abnormal. In offspring of men who had CVD before 50 years of age, seven different dyslipidemic profiles were present.1 Such results emphasize the importance of evaluating a lipoprotein profile in the fasting state. 
Table 166-3. Acceptable, Borderline, and High Plasma Lipid, Lipoprotein, and Apolipoprotein Concentrations (mg/dL) for Children and Adolescents*
 PRIMARY VERSUS SECONDARY DYSLIPOPROTEINEMIA
PRIMARY VERSUS SECONDARY DYSLIPOPROTEINEMIA
Before considering a dyslipoproteinemia to be a primary disorder, secondary causes must be excluded (Table 166-4). Each child with dyslipidemia should have routine blood tests to help rule out secondary causes of the disease. These include fasting blood sugar and tests of kidney, liver, and thyroid function. In secondary dyslipidemia, the associated disorder producing the dyslipidemia should be treated first in an attempt to normalize lipoprotein levels; however, if the dyslipidemia persists—for example, as it often does in type 1 diabetes and the nephrotic syndrome—the patient will require dietary treatment and, if indicated, drug therapy using the same guidelines as in primary dyslipidemias.
GUIDELINES FOR TREATING DYSLIPIDEMIA IN CHILDREN AND ADOLESCENTS
General guidelines for the dietary and pharmacological treatment of primary and secondary dyslipidemias in youth are presented here. Specific guidelines germane to each inherited disorder of dyslipidemia are provided as necessary in subsequent sections of this chapter.
 DIETARY THERAPY
DIETARY THERAPY
The first form of therapy for children with dyslipidemia is a diet containing decreased amounts of total fat, saturated fat, cholesterol, and simple sugars but containing increased complex carbohydrates. No decrease in total protein is recommended. Calories are sufficient to maintain normal growth and development. The NCEP pediatric panel recommended diet treatment after 2 years of age.5 Recent data from randomized clinical trials in general populations, such as STRIP, indicate that a diet low in total fat, saturated fat, and cholesterol may be instituted safely and effectively under medical supervision at 6 months of age.1
When to Initiate Treatment with Diet
If the first lipoprotein profile indicates that TC, LDL-C, non-HDL-C, or TG is elevated, or if the HDL-C is low (Table 166-3), then another confirmatory profile is obtained at least 3 weeks later. If dyslipidemia persists, secondary causes (Table 166-4) are ruled out and dietary treatment begun. A Step-One diet is usually started and the lipoprotein profile repeated in 6 to 8 weeks. If the dyslipidemia persists, then a Step-Two diet is initiated. Both diets require dietary counseling and physician monitoring. The Step-One diet calls for less than 10% of total calories from saturated fatty acids, no more than 30% of calories from total fat, and less than 300 mg/day of cholesterol. The Step-One diet is evaluated for at least 3 months before prescribing the Step-Two diet. The Step-Two diet entails further reduction of the saturated fatty acid intake to less than 7% of calories and reduced cholesterol intake to less than 200 mg/day.5
Safety and Efficacy of Dietary Therapy in Infants, Children, and Adolescents
The efficacy and safety of diets to treat dyslipidemia in youth have been demonstrated across the age spectrum of pediatric patients1—for example, from age 7 months to 7 years and from age 7 to age 11 in STRIP1 and from ages 8 to 10 throughout adolescence in the Dietary Intervention Study in Children (DISC).1 In some studies, there were lower intakes of calcium, zinc, vitamin E, and phosphorus on low-fat diets.1 Therefore, while normal growth is maintained on low-fat diets, attention needs to be paid to ensure adequate intake of these key nutritional elements. 
Human milk remains the gold standard for infant feeding.
Using margarines (about three servings daily) high in either plant stanol esters1 or plant sterol esters1 can reduce LDL-C an additional 10% to 15% when added to a low-fat diet. Water-soluble fibers such as psyllium can lower LDL-C an additional 5% to 10%.1
Consuming a soy protein beverage does not appear to lower LDL-C but does lower VLDL-C and TG and increases HDL-C.1 Garlic extract therapy does not lower LDL-C in hyper-lipidemic children.1
Garlic extract therapy does not lower LDL-C in hyper-lipidemic children.1
Overall, a diet low in fat for children with dyslipidemia appears both safe and efficacious. Medical and nutritional support is necessary to reinforce good dietary behaviors and to ensure nutritional adequacy. Human milk remains the gold standard for infant feeding.
Effect of a Low-Fat Diet in Childhood on Future CVD in Adulthood
That a low-saturated-fat, low-cholesterol diet in childhood will prevent CVD in adulthood can only be inferred from epidemiological studies.5 Obesity already promotes insulin resistance in childhood. In that regard, a low-saturated-fat dietary counseling program starting in infancy in STRIP improved insulin sensitivity in 9-year-old healthy children.1
 PHARMACOLOGICAL THERAPY
PHARMACOLOGICAL THERAPY
There are six main classes of lipid-altering drugs (Fig. 166-1): (1) inhibitors of HMG-CoA reductase (the statins), (2) bile acid sequestrants (BAS), (3) cholesterol absorption inhibitors (CAI), (4) niacin (nicotinic acid), (5) fish oils as omega-3 fatty acids (ecosapentanoic acid and decahexanoic acid), and (6) fibric acid derivatives.
Table 166-4. Causes of Secondary Dyslipidemia in Children and Adolescents



