Disorders of Heme Biosynthesis: The Porphyrias
Robert J. Desnick
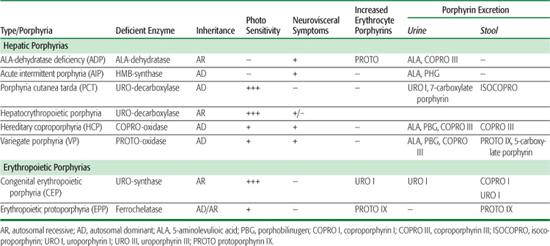
The porphyrias are a group of inherited and acquired metabolic disorders, each resulting from the deficient activity of a specific enzyme in the heme biosynthetic pathway.1,2 These enzyme deficiencies are inherited as autosomal dominant or recessive traits, with the exception of porphyria cutanea tarda (PCT), which usually is sporadic. These disorders are classified as either hepatic or erythropoietic, depending on the primary site of overproduction and accumulation of the porphyrin precursor(s) or porphyrin(s) (Table 167.1). Although some have overlapping features, manifestations of the hepatic porphyrias are neurological, including abdominal pain, neuropathy, and mental disturbances, whereas the erythropoietic porphyrias characteristically cause cutaneous photosensitivity. The neurological involvement in the hepatic porphyrias, which typically presents after puberty, results from the hepatic production of a neurotoxic metabolite, as liver transplantation ameliorated the frequent attacks in a patient with acute intermittent porphyria (AIP).3 Cutaneous sensitivity to sunlight may occur in infancy because of the excitation of excess porphyrins in the skin by long-wave ultraviolet light, which leads to cell damage, scarring, and deformation. Steroid hormones, drugs, and nutrition influence the production of porphyrin precursors and porphyrins, thereby precipitating or increasing the severity of some porphyrias.
Rare homozygous variants of the autosomal dominant hepatic porphyrias have been identified and usually manifest clinically before puberty. The symptoms in these patients are usually more severe and occur earlier than those of patients with the respective autosomal dominant porphyria (see below).1,4 Thus, the porphyrias are actually ecogenic disorders in which environmental, physiological, and genetic factors interact to cause disease.
Many symptoms of the porphyrias are nonspecific, and diagnosis is often delayed. Laboratory testing can confirm or exclude the diagnosis of a porphyria. Table 167.1 summarizes the major metabolites that accumulate in each porphyria. Urinary 5′-aminolevulinic acid (ALA) and porphobilinogen (PBG) are easily quantitated by chemical methods, and the urinary porphyrin isomers can be separated and quantitated by high-performance liquid chromatography. However, a definite diagnosis requires demonstration of the specific enzyme or gene defect. The isolation and characterization of the genes encoding all the heme biosynthetic enzymes have permitted the identification of the molecular lesions that cause each porphyria.1,2,5 Such molecular analyses make it possible to provide precise heterozygote or homozygote identification and prenatal diagnosis in families with known mutations.
The American Porphyria Foundation (www.porphyriafoundation.com) and the European Porphyria Initiative (www.porphyria-europe.org) sponsor informative and up-to-date Web sites. An extensive list of unsafe and safe drugs for individuals with porphyria is given at the Drug Database for Acute Porphyrias (www.drugs-porphyria.com).
Table 167-1 Clinical, Metabolic, and Genetic Characteristics of the Human Porphyrias
 HEME BIOSYNTHESIS
HEME BIOSYNTHESIS
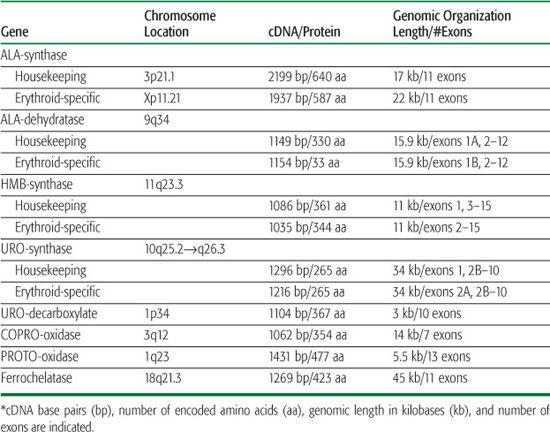
Heme biosynthesis involves eight enzymatic steps in the conversion of glycine and succinyl coenzyme A (CoA) to heme (Fig. 167-1 and Table 167.2). The first and last three enzymes in the heme biosynthetic pathway are located in the mitochondrion, whereas the other four are in the cytosol (see Fig. 167-1). The first enzyme, 5′-aminolevulinate synthase (ALA synthase), catalyzes the condensation of glycine, activated by pyridoxal phosphate and succinyl coenzyme A, to form ALA. This rate-limiting enzyme can be induced in the liver by a variety of drugs, steroids, and other chemicals. Distinct erythroid-specific and housekeeping forms of ALA synthase are encoded by separate genes located on chromosome 3p21.1 (ALAS1) and Xp11.2 (ALAS2). Defects in the X-linked erythroid gene ALAS2 cause X-linked sideroblastic anemia (see Chapter 168). 
 REGULATION OF HEME BIOSYNTHESIS
REGULATION OF HEME BIOSYNTHESIS
Regulation of heme synthesis differs in the two major heme-forming tissues, the liver and erythron. About 85% of the heme produced in the body is synthesized in erythroid cells to provide heme for hemoglobin; most of the remainder is produced in hepatocytes, primarily for synthesizing cytochromes. In the liver, the biosynthetic pathway is under negative feedback control. “Free” heme in the liver regulates the synthesis and mitochondrial translocation of the housekeeping form of ALA synthase (ALAS1). Heme represses the synthesis of the ALAS1 mRNA and interferes with the transport of the enzyme from the cytosol into mitochondria. Hepatic ALAS1 is inducible by many of the same chemicals that induce the cytochrome P450 enzymes in the liver’s endoplasmic reticulum. Because most of the heme in the liver is used for synthesizing cytochrome P450 enzymes, hepatic ALA synthase and the cytochrome P450 enzymes are regulated in a coordinated fashion.
Different regulatory mechanisms control production of heme for hemoglobin. The erythroid-specific ALA synthase (ALAS2) gene encoded on the X-chromosome is expressed at higher levels than the hepatic enzyme, and an erythroid-specific control mechanism regulates iron transport into erythroid cells. During erythroid differentiation, the activities of the heme biosynthetic enzymes are increased.
 THE HEPATIC PORPHYRIAS
THE HEPATIC PORPHYRIAS
The acute hepatic porphyrias are characterized by the rapid onset of neurological manifestations. During the acute attack, individuals have markedly elevated plasma and urinary concentrations of the porphyrin precursors ALA and PBG, which originate in the liver.
ALA DEHYDRATASE–DEFICIENT PORPHYRIA (ADP)
ADP is a rare autosomal recessive trait that has been described in only a few patients.7,8 Onset and severity of the disease are variable  . All ADP patients had significantly elevated levels of plasma and urinary ALA and markedly decreased ALA dehydratase activity. Treatment and prevention of neurological complications are the same as for other acute porphyrias.
. All ADP patients had significantly elevated levels of plasma and urinary ALA and markedly decreased ALA dehydratase activity. Treatment and prevention of neurological complications are the same as for other acute porphyrias.
 CLINICAL FEATURES
CLINICAL FEATURES
The first reported cases had clinical onset during adolescence of abdominal pain and neuropathy, resembling AIP. A Swedish infant presented with failure to thrive and required transfusions and parenteral nutrition. At age 63, a Belgian man developed an acute motor polyneuropathy that was associated with a myeloproliferative disorder. This patient was heterozygous for an ALA dehydratase mutation that presumably was present in erythroblasts, which underwent clonal expansion due to the bone marrow malignancy. 
 DIAGNOSIS
DIAGNOSIS
Patients have increased urinary levels of ALA and coproporphyrin. ALA dehydratase activity in erythrocytes is less than 5% of normal. Lead or succinylacetone (which accumulates in hereditary tyrosinemia and is structurally similar to ALA) can inhibit ALA dehydratase and increase urinary excretion. This may cause manifestations that resemble those of the acute porphyrias, lead intoxication (see Chapter 120) and hereditary tyrosinemia (fumarylacetoacetase deficiency; see Chapter 155). These conditions should therefore be considered in the differential diagnosis of ALA dehydratase–deficient porphyria. Immunologic studies in the reported cases demonstrated the presence of nonfunctional enzyme proteins that cross-reacted with anti-ALA–dehydratase antibodies. DNA analysis revealed different mutations (see the Human Gene Mutation Database, www.hgmd.org).
Heterozygotes are clinically asymptomatic and do not excrete increased levels of ALA, but they can be detected by demonstrating intermediate levels of erythrocyte ALA dehydratase activity or by demonstrating a specific mutation in the ALA dehydratase gene. Prenatal diagnosis of this disorder is possible  .
.
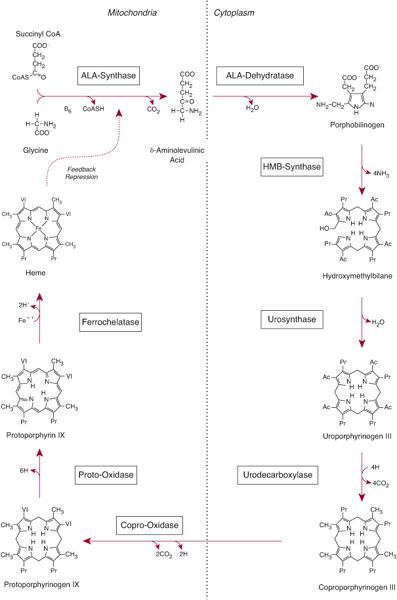
FIGURE 167-1. The human heme biosynthetic pathway.
 TREATMENT
TREATMENT
Treatment is similar to that of AIP.
ACUTE INTERMITTENT PORPHYRIA (AIP)
This hepatic porphyria is an autosomal dominant condition resulting from the half-normal level of HMB synthase (also termed PBG deaminase) activity.1,9 The disease is widespread but is especially common in Scandinavia and Great Britain. The enzyme deficiency can be demonstrated in most heterozygous individuals, but clinical expression is highly variable. Activation of the disease is related to ecogenic factors, such as drugs, diet, and steroid hormones, which can precipitate the manifestations. Attacks can be prevented by avoiding known precipitating factors. Rare cases of homozygous AIP have been reported in children.10
 CLINICAL FEATURES
CLINICAL FEATURES
Most heterozygotes remain clinically asymptomatic (latent) unless exposed to factors that increase porphyrin production. Endogenous and exogenous gonadal steroids, porphyrinogenic drugs, alcohol ingestion, and low-calorie diets (usually instituted for weight loss) are common precipitating factors. Table 167.3 lists the major drugs that are harmful in AIP (and in hereditary coproporphyria [HCP] and variegate porphyria [VP]) and some drugs and anesthetic agents known to be safe. More extensive lists of drugs considered harmful or safe are available at the Drug Database for Acute Porphyrias (www.drugs-porphyria.com) and at www.porphyriafoundation.com and www.porphyria-europe.com, but information is incomplete for many drugs. Attacks also can be provoked by infections and by surgery.
Because the neurovisceral symptoms rarely occur before puberty and are often nonspecific, a high index of suspicion is required to make the diagnosis. The disease can be disabling but is rarely fatal. Abdominal pain, the most common symptom, is usually steady and poorly localized but may be cramping. Ileus, abdominal distention, and decreased bowel sounds are common. However, increased bowel sounds and diarrhea may occur. Abdominal tenderness, fever, and leukocytosis are usually absent or mild, because the symptoms are neurological rather than inflammatory. Nausea; vomiting; constipation; tachycardia; hypertension; mental symptoms; muscle weakness; sensory loss; dysuria; urinary retention; and pain in the limbs, head, neck, or chest are characteristic. Tachycardia, hypertension, restlessness, tremors, and excess sweating are due to sympathetic overactivity.
The peripheral neuropathy is the result of axonal degeneration (rather than demyelination) and affects primarily motor neurons. Significant neuropathy does not occur with all acute attacks; abdominal symptoms are usually more prominent. Motor neuropathy initially affects the proximal muscles, more often in the shoulders and arms. The course and degree of involvement are variable. Deep-tendon reflexes may be normal or hyperactive but are usually decreased or absent with advanced neuropathy. Motor weakness can be asymmetric and focal and may involve cranial nerves. Sensory changes such as paresthesias and loss of sensation are less prominent. Progressive muscle weakness can lead to respiratory and bulbar paralysis and death when diagnosis and treatment are delayed. Sudden death may result from sympathetic overactivity and cardiac arrhythmia.
Mental symptoms, such as anxiety, insomnia, depression, disorientation, hallucinations, and paranoia, can occur in acute attacks. Seizures can be caused by neurological effects or by hyponatremia. Treatment of seizures is difficult, because virtually all antiseizure drugs (except bromides) may exacerbate AIP (clonazepam may be safer than phenytoin or barbiturates). Hyponatremia results from hypothalamic involvement and inappropriate secretion of antidiuretic hormone, or from electrolyte depletion due to vomiting, diarrhea, poor intake, or excess renal sodium loss. Persistent hypertension and impaired renal function may occur. When an attack resolves, abdominal pain may disappear within hours, and paresis begins to improve within days and may continue to improve over several years.
Table 167-2 The Human Heme Biosynthesis Genes*
 DIAGNOSIS
DIAGNOSIS
ALA and PBG levels are increased in plasma and urine during acute attacks.9 Although the diagnosis of an acute attack is based on clinical findings and not the absolute level of these porphyrin precursors, the increase is expected to be substantial. PBG excretion is usually 50 to 200 mg/24 hr, and urinary ALA excretion is 20 to 100 mg/24 hr. The excretion of these compounds generally decreases with clinical improvement, particularly after hematin infusions. A normal urinary PBG level effectively excludes AIP as a cause for current symptoms. Fecal porphyrins are usually normal or minimally increased in AIP, in contrast to HCP and VP. Most asymptomatic (“latent”) heterozygotes with HMB synthase deficiency have normal urinary excretion of ALA and PBG. Therefore, measuring HMB synthase in erythrocytes may be useful to confirm the diagnosis and to screen asymptomatic family members. 
Table 167-3 Categories of Unsafe and Safe Drugs in AIP, HCP, and VP

The enzyme deficiency is detectable in erythrocytes from most AIP heterozygotes (classic AIP). Over 250 mutations causing AIP have been identified (see the Human Gene Mutation Database, www.hgmd.org). 
Identifying the gene in an index case enables detection of latent family members and prenatal diagnosis of an at-risk fetus using cultured amniotic cells or chorionic villi. However, this is seldom done, because the prognosis of individuals with HMB synthase mutations is generally favorable.
 TREATMENT
TREATMENT
During acute attacks, narcotic analgesics are usually required for abdominal pain, and phenothiazines are useful for nausea, vomiting, anxiety, and restlessness.9 Chloral hydrate can be given for insomnia, and benzodiazepines in low doses are probably safe if a minor tranquilizer is required. Carbohydrate loading, usually with intravenous glucose (at least 300 g/dL), may be effective in milder acute attacks of porphyria (without paresis, hyponatremia, etc). Because intravenous hemin is more effective and the response slower if treatment is delayed, it is no longer recommended that hemin therapy for a severe attack be started only after an unsuccessful trial of intravenous glucose for several days. Hemin should be used initially for severe attacks and for mild attacks that do not respond to carbohydrate loading within 1 to 2 days. The standard regimen is 3 to 4 mg of heme in the form of lyophilized hematin (Ovation Pharmaceuticals), heme albumin (hematin reconstituted with human albumin) or heme arginate (Orphan Europe), infused daily for 4 days.9,11 Heme arginate and heme albumin are chemically stable and are less likely than hematin to produce phlebitis or an anticoagulant effect. The rate of recovery from an acute attack depends on the degree of neuronal damage and may be rapid (1 to 2 days) with prompt therapy. Recovery from severe motor neuropathy may continue for months or years. 
An allogeneic liver transplant was performed on a 19-year-old female AIP heterozygote who had 37 acute attacks in the 29 months prior to transplantation.3 Post-transplantation, her elevated urinary ALA and PBG levels returned to normal in 24 hours, and she did not experience acute neurological attacks for more than 18 months. Liver transplantation is a high-risk procedure and should not be considered as an established treatment for acute porphyrias.
 HOMOZYGOUS DOMINANT AIP
HOMOZYGOUS DOMINANT AIP
Homozygous dominant AIP is a rare form of porphyria presenting in infancy. Patients inherit HMB synthase mutations from each of their heterozygous parents; therefore, they have very low (< 2%) enzyme activity.10 In these homozygous-affected patients, disease manifestations included failure to thrive, developmental delay, bilateral cataracts, or hepatosplenomegaly. Acute attacks did not occur. Urinary ALA and PBG were markedly elevated. Studies of brain MRIs of children with homozygous AIP have suggested damage primarily in white matter that was myelinated postnatally, while tracks that myelinated prenatally were normal.10 These findings suggest that a neurotoxic endogenous product, such as ALA or PBG, present in large amounts postnatally (rather than heme deficiency) caused nervous tissue damage. Prenatally, excess amounts of ALA and PBG cross the placenta and are excreted in the mother’s urine. Most children with homozygous AIP die at an early age.
PORPHYRIA CUTANEA TARDA (PCT)
Porphyria cutanea tarda (PCT), the most common of the porphyrias, can be sporadic (type I) or familial (types II and III). Hepatic URO decarboxylase is deficient in all types of PCT and for clinical manifestations must be substantial (< 20% of normal). In type I PCT, URO decarboxylase activity is normal in erythrocytes. In type II PCT, an autosomal dominant trait, the enzyme is systemically deficient. In type III PCT, which clusters in families, deficiency of the enzyme activity is limited to the liver. Hepatoerythropoietic porphyria (HEP) is an autosomal recessive form of porphyria, as affected individuals inherited an URO decarboxylase mutation from each parent and have severe systematic deficiency of URO decarboxylase activity. HEP usually presents in childhood. 
 CLINICAL FEATURES
CLINICAL FEATURES
Cutaneous photosensitivity is the major clinical feature. Neurological manifestations are not observed. Other features include hypertrichosis and hyperpigmentation, especially of the face, and thickening, scarring, and calcification resembling the cutaneous changes of systemic sclerosis. 
Hepatoerythropoietic porphyria (HEP) resembles congenital erythropoietic porphyria (CEP) and usually presents with blistering skin lesions, hypertrichosis, scarring, and red urine in infancy or childhood.
 DIAGNOSIS
DIAGNOSIS
Porphyrin levels are increased in the liver, plasma, urine, and stool. The urinary ALA level may be slightly increased, but the PBG level is normal. Urinary porphyrins consist mostly of uroporphyrin and 7-carboxylate porphyrin, with lesser amounts of coproporphyrin and 5- and 6-carboxylate porphyrins. Plasma porphyrins also are increased in a pattern that resembles that in urine. Isocoproporphyrins are increased in feces and sometimes in plasma and urine. The finding of increased isocoproporphyrins is diagnostic for a deficiency of hepatic URO decarboxylase.
Type II PCT and HEP can be diagnosed by finding decreased URO decarboxylase activity in erythrocytes or by indentifying mutations in the URO decarboxylase gene. URO decarboxylase activity in liver, erythrocytes, and cultured skin fibroblasts in type II PCT is approximately 50% of normal in affected individuals and in family members with latent disease. In HEP, the URO decarboxylase activity is markedly deficient, with typical levels of 3% to 10% of normal.
 TREATMENMT
TREATMENMT
A complete response can almost always be achieved by repeated phlebotomy to reduce hepatic iron. Because iron overload is not marked in most cases, remission may occur after only five or six phlebotomies. PCT patients with hemochromatosis may require many more phlebotomies. After remission, continued phlebotomy may not be needed, even if ferritin levels return to normal. Relapses are treated again by phlebotomy. 
PCT also can be treated with chloroquine or hydroxychloroquine, both of which complex with the excess porphyrins and promote their excretion. Small doses (eg, 125 mg chloroquine phosphate twice weekly) should be given, because standard doses can induce transient, sometimes marked, increases in photosensitivity and hepatocellular damage. Hepatic imaging can diagnose or exclude complicating hepatocellular carcinoma. Treating PCT in patients with end-stage renal disease is facilitated by administering erythropoietin.
 HEPATOERYTHROPOIETIC PORPHYRIA (HEP)
HEPATOERYTHROPOIETIC PORPHYRIA (HEP)
Hepatoerythropoietic porphyria (HEP), which is the homozygous form of familial (type 2) PCT, resembles CEP clinically.4 Excess porphyrins originate mostly from the liver, with a pattern consistent with severe URO decarboxylase deficiency. There also is a substantial increase in erythrocyte zinc protoporphyrin in HEP, as in homozygous dominant forms of the acute porphyrias, ADP, and some cases of CEP.
HEP usually presents with blistering skin lesions, hypertrichosis, scarring, and red urine in infancy or childhood. Sclerodermoid skin changes are sometimes prominent. 
HEP is readily distinguished from CEP by increases in both uroporphyrin and heptacarboxyl porphyrin in urine and in isocoproporphyrins in stool. 
HEREDITARY COPROPORPHYRIA (HCP)
Hereditary coproporphyria (HCP) is an autosomal dominant hepatic porphyria that results from half-normal levels of COPRO oxidase activity. Photosensitivity may occur. Cases of homozygous dominant HCP have been reported.
 CLINICAL FEATURES
CLINICAL FEATURES
HCP is influenced by the same factors that cause attacks in AIP. The disease is latent before puberty, and symptoms are more common in women. Neurovisceral symptoms and other manifestations are virtually identical to those of AIP. Photosensitivity may resemble that in PCT and VP. Cutaneous lesions may begin in childhood in the rare homozygous dominant cases.
 HOMOZYGOUS DOMINANT HCP AND HARDEROPORPHYRIA
HOMOZYGOUS DOMINANT HCP AND HARDEROPORPHYRIA
Individuals with mutations in both their COPRO oxidase alleles and markedly decreased COPRO oxidase activities have been described; these mutations cause homozygous dominant HCP or a rare variant form called harderoporphyria.4 Homozygous HCP presents early in childhood with symptoms of growth retardation, hypertrichosis, and skin hyperpigmentation.1,4 Later, these patients can have acute porphyric attacks.
Individuals with harderoporphyria, which is a biochemical and clinical variant form of HCP in which hemolysis and erythropoietic features are prominent, usually present in early childhood with jaundice, hemolytic anemia, hepatosplenomegaly, and skin photosensitivity.1,4 However, the symptoms may be variable; acute attacks do not occur.
 DIAGNOSIS
DIAGNOSIS
Coproporphyrin concentrations are markedly increased in the urine and feces of HCP patients when the disease is symptomatic and sometimes when there are no symptoms. Urinary ALA and PBG levels are increased during acute attacks but may return to normal when symptoms resolve. Although the diagnosis can be confirmed by measuring COPRO oxidase activity, these assays are not widely available and require cells other than erythrocytes. COPRO oxidase gene mutations are diagnostic, and over 25 have been reported (see the Human Gene Mutation Database, www.hgmd.org). Patients with homozygous HCP have markedly increased fecal coproporphyrin III due to markedly deficient (<10%) COPRO oxidase activity. In patients with harderoporphyria, urinary coprotoporphyrin III, fecal porphyrins (66–90% harderoporphyrin), and zinc protoporphyrin are increased. All harderoporphyria patients reported to date are either homoallelic or heteroallelic for the K404E missense mutation.
 TREATMENT
TREATMENT
Neurological symptoms are treated as in AIP. Phlebotomy and chloroquine are ineffective when cutaneous lesions are present.
VARIEGATE PORPHYRIA (VP)
Variegate porphyria (VP), a hepatic porphyria that results from the deficient activity of PROTO oxidase, is inherited as an autosomal dominant trait and can present with neurological symptoms, photosensitivity, or both. Homozygous VP is rare and presents in early childhood.1,4
 CLINICAL FEATURES
CLINICAL FEATURES
Neurovisceral signs and symptoms develop after puberty and are similar to those of AIP or HCP. 
 HOMOZYGOUS-DOMINANT VP
HOMOZYGOUS-DOMINANT VP
Affected individuals with homozygous-dominant VP have mutations affecting both PROTO oxidase alleles, resulting in very low enzyme activity levels.1,4,14 These patients generally develop cutaneous symptoms, including photosensitivity and hypertrichosis, before the age of 2 years. Scarring and deformities of the face and digits may be prominent. Most patients do not have acute attacks. Neurological symptoms in some patients include mental retardation, convulsions, growth retardation, and nystagmus. Laboratory findings include elevated erythrocyte zinc protoporphyrin levels, as in other homozygous-dominant porphyrias. Mutations have been identified in most homozygous VP patients. 
 DIAGNOSIS
DIAGNOSIS
Urinary ALA and PBG levels are increased during acute attacks but may return to normal more quickly than in AIP. Increases in fecal protoporphyrin and coproporphyrin III and in urinary coproporphyrin III are more persistent. Plasma levels of porphyrins are increased, particularly when there are cutaneous lesions. VP can be distinguished rapidly from all other porphyrias by examining the fluorescence emission spectrum of porphyrins in plasma at neutral pH. This test is particularly useful for differentiating VP from PCT. 
 TREATMENT
TREATMENT
As in AIP, acute attacks are treated with hematin. Other than avoiding sun exposure, there are few effective measures for treating the skin lesions. Beta-carotene, phlebotomy, and chloroquine are not helpful.
 THE ERYTHROPOIETIC PORPHYRIAS
THE ERYTHROPOIETIC PORPHYRIAS
In the erythropoietic porphyrias, porphyrins from bone marrow erythrocytes and plasma are deposited in the skin and lead to cutaneous photosensitivity.
CONGENITAL ERYTHROPOIETIC PORPHYRIA (CEP)
Congenital erythropoietic porphyria (CEP) is an autosomal recessive disorder, also known as Gunther disease, that results from the markedly deficient activity of URO synthase and is associated with hemolytic anemia and severe cutaneous photosensitivity.1,15 CEP is characterized by accumulation of uroporphyrin I and coproporphyrin I isomers.
 CLINICAL FEATURES
CLINICAL FEATURES
Severe cutaneous photosensitivity begins in early infancy. The disease may be recognized in utero as a cause of nonimmune hydrops fetalis. The skin over sun-exposed areas is friable, and bullae and vesicles are prone to rupture and infection. Skin thickening, focal hypo- and hyperpigmentation, and hypertrichosis of the face and extremities are characteristic. Secondary infection of the cutaneous lesions can disfigure the face and hands. Porphyrins are deposited in teeth and bones. Consequently, the teeth are reddish brown and fluoresce on exposure to long-wave ultraviolet light. Hemolysis is probably due to the marked increase in erythrocyte porphyrins and leads to splenomegaly. Adults with a milder form of the disease have been described.
 DIAGNOSIS
DIAGNOSIS
Uroporphyrin and coproporphyrin (mostly type I isomers) accumulate in the bone marrow, erythrocytes, plasma, urine, and feces. The diagnosis should be confirmed by demonstration of markedly deficient URO synthase activity and/or URO synthase gene mutations.15,16 The disease can be detected in utero by measuring porphyrins in amniotic fluid and URO synthase activity in cultured amniotic cells or chorionic villi. Over 35 URO synthase mutations have been reported (see the Human Gene Mutation Database, www.hgmd.org).
 TREATMENT
TREATMENT
The transfusion of sufficient blood to suppress erythropoiesis is effective but results in iron overload. Splenectomy may reduce hemolysis and decrease transfusion requirements. Protection from sunlight is essential, and minor skin trauma should be avoided. Beta-carotene may be of some value. Complicating bacterial infections should be treated promptly. Recently, bone marrow or cord-blood transplantation has proven effective in several transfusion-dependent children, providing the rationale for stem-cell gene therapy.
ERYTHROPOIETIC PROTOPORPHYRIA (EPP)
Erythropoietic protoporphyria (EPP) is an inherited disorder resulting from the markedly deficient activity of FECH activity, the last enzyme in the heme biosynthetic pathway. EPP is the most common erythropoietic porphyria and is the most common porphyria in children and the second most common in adults. EPP patients have FECH activities as low as 15% to 25% in lymphocytes and cultured fibroblasts. In most patients, a disabling (ie, causative) mutation in one FECH allele is combined with a relatively common intron 3 (IVS3) single nucleotide alteration (IVS3-48T → C) in the other allele, which results in decreased amounts of the normal enzyme. 
 CLINICAL FEATURES
CLINICAL FEATURES
Skin photosensitivity usually begins in childhood. The skin manifestations differ from those of other porphyrias. Vesicular lesions are uncommon. Redness, swelling, burning, and itching can develop within minutes of sun exposure and can resemble angioedema. Symptoms may seem out of proportion to the visible skin lesions. Sparse vesicles and bullae occur in 10% of cases. Chronic skin changes may include lichenification, leathery pseudovesicles, labial grooving, and nail changes. Severe scarring is rare, as are pigment changes, friability, and hirsutism.
The primary source of excess protoporphyrin is the bone marrow reticulocyte. Erythrocyte protoporphyrin is free (not complexed with zinc) and is mostly bound to hemoglobin. In plasma, protoporphyrin is bound to albumin. Hemolysis and anemia are usually absent or mild.
Liver function is usually normal, but up to 20% of EPP patients may have minor abnormalities of liver function, and in some patients (∼5%), accumulation of protoporphyrin causes chronic liver disease that can progress to liver failure and death. The hepatic complications are often preceded by increasing levels of erythrocyte and plasma protoporphyrin and probably result, in part, from protoporphyrin accumulation in the liver. Protoporphyrin is insoluble; it forms crystalline structures in liver cells and can decrease hepatic bile flow. Some patients have gallstones that are at least partially composed of protoporphyrin.
 DIAGNOSIS
DIAGNOSIS
A substantial increase in erythrocyte protoporphyrin, which is predominantly free and not complexed with zinc, is the hallmark of this disease. Protoporphyrin levels are also variably increased in bone marrow, plasma, bile, and feces. Plasma and fecal porphyrins are less increased than in most other cutaneous porphyrias and are sometimes normal. Therefore, measuring erythrocyte protoporphyrin is important for diagnosis. FECH mutation analysis is recommended to detect the causative mutation and, in most affected families, the presence of the IVS3-48T →C alteration in the other FECH allele. 
 TREATMENT
TREATMENT
Avoiding sunlight exposure and wearing clothing designed to provide protection for individuals with chronic photosensitivity are essential. Oral beta-carotene improves tolerance to sunlight in many patients. 
Treating hepatic complications is difficult. Liver transplantation has been performed in some patients with severe liver complications. However, liver disease often eventually recurs in the transplanted liver because of continued bone marrow production of excess protoporphyrin. 
ACKNOWLEDGMENTS
This work was supported in part by grants from the National Institutes of Health, including a research grant (5 R01 DK026824); the General Clinical Research Center Programs at the Mount Sinai School of Medicine (5 M01 RR00071); and the National Center for Research Resources.
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


