Disorders of Glucose Transporters
René Santer
GENERAL CONSIDERATIONS
D-glucose and other monosaccharides are hy-drophilic substances that cannot easily cross the lipophilic bilayer of the cell membrane. Since carbohydrates are most important for supplying energy to essentially all cell types, specific transport mechanisms have evolved. While vesicle-associated glucose transport has been described only recently,1-4 transporter proteins have been known for years. Such proteins are embedded into the cell membrane and function as hydrophilic pores that allow cellular uptake and release and allow transcel-lular transport of monosaccharides.
Glucose transporter proteins can be divided into two groups: (1) Sodium-dependent glucose transporters (SGLTs, symporter systems, secondary “active” transporters), which are members of the solute carrier family 5 (SLC5), couple sugar transport to the electrochemical gradient of sodium and hence can transport glucose against its own concentration gradient (eFig. 157.1  ); and (2) facilitative glucose transporters (GLUTs, uniporter systems, “passive” transporters) are members of the SLC2 family that can transport monosaccharides only along an existing gradient (eFig. 157.2
); and (2) facilitative glucose transporters (GLUTs, uniporter systems, “passive” transporters) are members of the SLC2 family that can transport monosaccharides only along an existing gradient (eFig. 157.2  ). To date, five congenital defects of monosaccharide transport are known (Fig. 157-1). Their clinical picture is the consequence of tissue-specific expression and substrate specificity of the affected transporter.
). To date, five congenital defects of monosaccharide transport are known (Fig. 157-1). Their clinical picture is the consequence of tissue-specific expression and substrate specificity of the affected transporter. 
CONGENITAL GLUCOSE-GALACTOSE MALABSORPTION
 PATHOPHYSIOLOGY AND GENETICS
PATHOPHYSIOLOGY AND GENETICS
A congenital defect of the sodium-dependent monosaccharide transporter SGLT1 at the apical membrane of enterocytes is the basic defect of congenital glucose-galactose malabsorption (GGM; OMIM 182380).5
Malabsorption of the two monosaccharides results in osmotic effects with massive fluid loss into the intestine.  Both truncating and missense mutations of the SGLT1 gene SLC5A1 have resulted in the absence of a functioning transporter protein within the apical enterocyte membrane.12 The fact that patients with GGM show mild glucosuria points to a physiological role of this transporter in renal glucose reabsorption.13
Both truncating and missense mutations of the SGLT1 gene SLC5A1 have resulted in the absence of a functioning transporter protein within the apical enterocyte membrane.12 The fact that patients with GGM show mild glucosuria points to a physiological role of this transporter in renal glucose reabsorption.13
GGM is a relatively rare autosomal recessive disorder, but the exact prevalence is unknown. SLC5A1, located on chromosome 22q13, comprises 15 exons and codes for a protein of 664 amino acids that forms 14 transmembraneous loops.9 To date, approximately 60 different mutations have been found5,12 scattered all over the gene; the existence of a mutational hot spot is controversial.
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
In general, children with GGM present within days after a normal pregnancy and birth. First clinical signs are bloating and profuse watery diarrhea. Stools are so loose that they may be mistaken for urine. Typically, polyhydramnios is not observed. Both breast- and bottle-fed infants are affected, and symptoms may even begin when newborns are given only tea sweetened with glucose or polymers of glucose. As a result, patients develop severe hypertonic dehydration with fever, which may be misinterpreted as a sign of an intestinal infection. If the correct diagnosis is missed and glucose and galactose are not eliminated from the diet, and if parenteral fluid administration is not available, patients die from hypovolemic shock. In typical cases, the diagnosis is considered after repeated frustrating attempts to switch from parenteral fluids to oral feeds.14 Chronic dehydration might be responsible for nephrolithiasis and nephrocalcinosis that develop in many cases.15
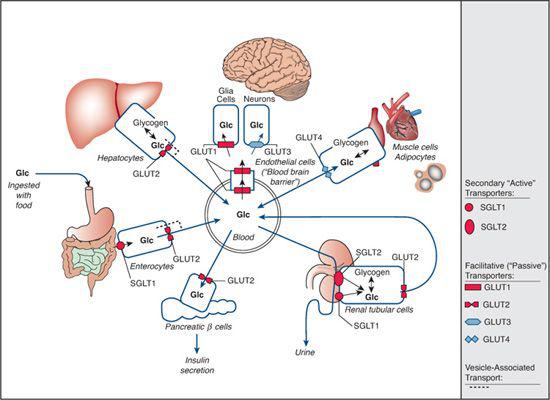
FIGURE 157-1. Overview of glucose transporters within the human organism. Transport across cell membranes is depicted by arrows, and specific transporters are shown as symbols: rounded symbols are used for sodium-dependent, secondary “active” transporters (SGLTs); angular symbols are used for facilitative, “passive” transporters (GLUTs). Red symbols represent the known defects of SGLT1 (glucose-galactose malabsorption), SGLT2 (familial renal glucosuria), GLUT1 (glucose transporter-1 deficiency), and GLUT2 (Fanconi-Bickel syndrome). GLUT10 deficiency (arterial tortuosity syndrome) is not depicted here (see text for more details).
 DIAGNOSTIC EVALUATION
DIAGNOSTIC EVALUATION
Due to its life-threatening character, GGM must be suspected clinically and treatment must be initiated before an ultimate diagnosis is established. The clinical stabilization on parenteral nutrition with no foods given per mouth or on a fructose-based formula are in favor of the diagnosis.  Although costly, molecular genetic studies on genomic DNA are recommended early if clinical signs are suggestive, particularly if prenatal diagnosis is likely to be requested in a future pregnancy.16
Although costly, molecular genetic studies on genomic DNA are recommended early if clinical signs are suggestive, particularly if prenatal diagnosis is likely to be requested in a future pregnancy.16
 TREATMENT AND PROGNOSIS
TREATMENT AND PROGNOSIS
Whenever GGM is considered, glucose and galactose should be omitted from the diet. A formula containing fructose as the only carbohydrate is easily prepared by adding monosaccharide to commercially available carbohydrate-free dietary products. The preparation of the diet becomes more complicated when additional foods are introduced, but glucose tolerance improves with age by an as yet unknown mechanism.17
RENAL GLUCOSURIA
 PATHOPHYSIOLOGY AND GENETICS
PATHOPHYSIOLOGY AND GENETICS
Renal glucosuria (OMIM 233100) is an isolated defect of tubular glucose reabsorption at the proximal tubules and does not affect any other glomerular or tubular kidney functions.18 A large number of individuals with renal glucosuria carry mutations within the SGLT2 gene SLC5A2,19-21 located on chromosome 16p11.22 Its product is a low-affinity carrier that transports glucose (but not galactose) across the apical membrane of proximal renal tubulus cells. 
Homozygosity or compound heterozygosity for SLC5A2 mutations results in the severe types of renal glucosuria, generally in the range of 10 to more than 100 g/1.73 m2/d, whereas heterozygosity is associated with milder glucosuria of 0.4-5.0 (up to 10) g/1.73 m2/d. Since glucosuria has not been observed constantly in all heterozygous carriers of mutations,20 inheritance of renal glucosuria is best characterized as a codominant trait with variable penetrance. To date, approximately 50 SGLT2 mutations have been described, which are scattered all over the gene. Most of them affect only single pedigrees; only a splice mutation was found in several kindreds of different ethnic backgrounds.20,21
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Mild renal glucosuria, generally considered a “nondisease,” is relatively common. Individuals with a higher glucose excretion or even virtual absence of tubular glucose reabsorption (termed renal glucosuria type 0) are extremely rare. Only a small number of patients present with polyuria or enuresis. Most individuals with renal glucosuria are detected during a routine urine examination when a positive urine test for glucose is found. All other tubular functions are normal; there are no signs of renal tubular acidosis, of rickets due to tubular phosphate loss, and so on. Also, mild hyperaminoaciduria is generally not present but can occasionally be a secondary phenomenon in individuals with massive glucosuria.23 Renal glucosuria is an important differential diagnosis to diabetes mellitus, which can be easily excluded by the presence of normal blood glucose concentrations. 
 DIAGNOSTIC EVALUATION
DIAGNOSTIC EVALUATION
Diagnosis is straightforward in patients with glucosuria and normoglycemia who do not show any other evidence of renal tubular dysfunction.
 TREATMENT AND PROGNOSIS
TREATMENT AND PROGNOSIS
For most cases, dietary treatment is not necessary, and prognosis, even in individuals with type 0 glucosuria, is excellent.24
GLUCOSE TRANSPORTER-1 DEFICIENCY SYNDROME
 PATHOPHYSIOLOGY AND GENETICS
PATHOPHYSIOLOGY AND GENETICS
Glucose transporter (GLUT)1 is a membrane-spanning, glycosylated protein that provides basal glucose entry across most blood-tissue barriers. This protein exclusively facilitates glucose transport across the luminal and abluminal membranes of brain capillaries that represent the blood-brain barrier created by the impermeable tight junctions between these cells. Consequently, GLUT1 deficiency (OMIM 606777) results in a low glucose concentration in the cerebrospinal fluid, termed hypoglycorrhachia. Thus, GLUT1 is important for glucose supply to glial cells and neurons.26
The majority of patients with GLUT1 deficiency carry heterozygous de novo mutations in the GLUT1 gene SLC2A1, which is located on chromosome 1p. Mutations are distributed randomly and are of various types (missense, nonsense, splice-site mutations, and large deletions).28 Phenotype-genotype correlation is yet unclear, but missense mutations may cause a milder phenotype than haploinsufficiency. Autosomal dominant GLUT1 deficiency has been identified in a few unrelated families.28
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Glucose transporter (GLUT)1 deficiency syndrome typically presents as an early onset epileptic encephalopathy. For unknown reasons, fetal development is undisturbed; pregnancy, delivery, and the neonatal period are uneventful. The majority of patients develop epilepsy within the first year of life as cerebral glucose demand increases. Occasionally, patients without epilepsy have been identified29; others develop only paroxysmal dyskinesia. Seizures are of various types and frequency, are often refractory to anticon-vulsants, and are sometimes aggravated by fasting. In infants, peculiar eye movements, staring spells, drop attacks, and cyanotic spells are the most frequent symptoms. Older children present predominantly with epilepsy absences, myoclonus, and grand mal seizures.30 Interictal EEGs in GLUT1 deficiency may be normal regardless of age. Occasionally, EEG recordings may show an improvement after glucose intake.  No structural brain abnormalities are detected at neuroimaging, but PET studies may show a diminished cortical uptake with a more severe reduction in metabolism in the mesial temporal regions and thalami, accentuating a relative signal increase in the basal ganglia.32
No structural brain abnormalities are detected at neuroimaging, but PET studies may show a diminished cortical uptake with a more severe reduction in metabolism in the mesial temporal regions and thalami, accentuating a relative signal increase in the basal ganglia.32
No dysmorphic features are observed. In early childhood, global developmental delay and a complex motor disorder become apparent. Motor milestones are delayed, and speech is often significantly slurred and slowed, but all patients acquire speech and mobility. Hypotonia and ataxia result in a broad-based, unsteady gait. A substantial number of patients display additional dystonic features and elements of spasticity. Patients are of a friendly disposition and continue to make developmental progress. GLUT1 deficiency is not a degenerative disorder, though individuals with severe cases develop secondary microcephaly.30,33
 DIAGNOSTIC EVALUATION
DIAGNOSTIC EVALUATION
GLUT1 deficiency should be suspected in any patient with hypoglycorrhachia if hypoglyce-mia or a central nervous system (CNS) infection are absent. In affected individuals, absolute values for cerebrospinal fluid (CSF) glucose concentrations are generally less than 45 mg/dl, but for diagnosis, the CSF-to-blood-glucose ratio is superior to the absolute concentration. This should be obtained in a nonictal, metabolic steady state following a 4- to 6-hour fast with blood glucose determined before the lumbar puncture to avoid stress-related hyperglyce-mia. Hypoglycorrhachia with a ratio less than 0.46 together with a low to normal CSF lactate is diagnostic  .
.
GLUT1 deficiency should be confirmed by molecular genetic methods or by glucose uptake studies in erythrocytes (in which 5% of their membrane protein is GLUT1). In such studies, glucose uptake is reduced to about half of the control values.34
 TREATMENT AND PROGNOSIS
TREATMENT AND PROGNOSIS
GLUT1 deficiency syndrome can be successfully treated with a high-fat, low-carbohydrate (“ke-togenic”) diet.35,36 Ketone bodies derived from dietary fat restore brain energy metabolism since the transport route into the CNS is not dependent on GLUT1. Such a diet can effectively control seizures, but anticonvulsant medication may be necessary in the later course of the disease, and neurological function and movement disorder usually remain impaired.33 In contrast to intractable epilepsy, for which such a diet was originally suggested, it is recommended that patients with GLUT1 deficiency remain on a ketogenic diet throughout childhood and into adolescence, by which time the cerebral glucose demands decrease to adult levels. 
FANCONI-BICKEL SYNDROME
 PATHOPHYSIOLOGY, GENETICS
PATHOPHYSIOLOGY, GENETICS
Fanconi-Bickel syndrome (FBS; OMIM 227810) is caused by congenital deficiency or impaired function of glucose transporter (GLUT)2, a high Km monosaccharide carrier that transports glucose and galactose.38,39 Main clinical features of FBS can be explained by the fact that this facilitative carrier is expressed both in hepatocytes and at the basolateral membrane of reabsorbing cells of the proximal tubules. 
In hepatocytes, GLUT2 seems to function as a glucose sensor. In the fasting state, when extracellular glucose concentration declines, the concentrations of glucose and glucose-6-phosphate within hepatocytes are inappropriately high in FBS patients. This stimulates glycogen synthesis and inhibits gluconeogenesis and glycogenolysis, which ultimately predisposes the individual to hypoglycemia and hepatic glycogen accumulation.39 Impaired transport of glucose out of renal tubular cells results in the accumulation of glycogen and free glucose within these cells. This impairs other transport functions and results in a generalized tubulopathy with disproportionately severe glucosuria. The extreme amounts of glucose lost with the urine (even at times when blood glucose is low) may contribute to the propensity to develop hypoglycemia.
FBS is a very rare autosomal recessive condition caused by mutations of the GLUT2 gene SLC2A2.  The human gene, mapped to chromosome 3q26, codes for a 524 amino acid protein with 55% amino acid identity to GLUT1. In contrast to sodium-dependent glucose transporters (SGLTs), all GLUT proteins form 12 transmembranous loops within the cell membrane. The genomic structure of SLC2A2 encompasses 11 exons,41 and to date, approximately 70 different mutations scattered throughout the gene have been detected.39,40
The human gene, mapped to chromosome 3q26, codes for a 524 amino acid protein with 55% amino acid identity to GLUT1. In contrast to sodium-dependent glucose transporters (SGLTs), all GLUT proteins form 12 transmembranous loops within the cell membrane. The genomic structure of SLC2A2 encompasses 11 exons,41 and to date, approximately 70 different mutations scattered throughout the gene have been detected.39,40
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
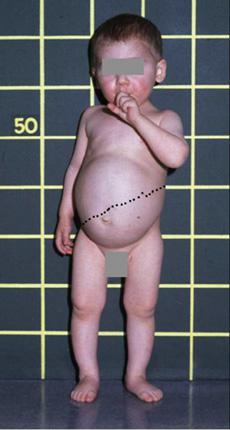
Patients with Fanconi-Bickel syndrome (FBS) typically present at an age of 3 to 10 months with hepatomegaly, a Fanconi-type nephropathy, a propensity to hypoglycemia in the fasting state, and glucose and galactose intolerance in the fed state.39,42 A few cases have presented during neonatal screening with hypergalactosemia,43 and cataracts have occasionally been observed in infants with FBS.44 At an early stage, hepatomegaly, which is caused by massive accumulation of glycogen, may not yet be present, and nonspecific symptoms such as fever, vomiting, chronic diarrhea, and failure to thrive may predominate. With increasing age, the clinical presentation of protuberant abdomen, moon-shaped face, and short stature becomes more and more similar to other hepatic glycogen storage diseases (Fig. 157-2). Also, the kidneys accumulate glycogen, and their enlargement can be detected by ultrasound. Hypophosphatemic rickets is the major manifestation of tubular dysfunction, which results in joint swelling, bowing of legs, and pathological fractures. FBS patients have an entirely normal mental development, but growth and puberty are severely retarded.39,42
 DIAGNOSTIC EVALUATION
DIAGNOSTIC EVALUATION
Diagnosis of FBS is suggested by the characteristic combination of an altered glucose homeostasis, a hepatic glycogen accumulation, and the typical features of a Fanconi-type tubulopathy. Fasting hypoglycemia and impaired glucose and galactose tolerance may be documented during oral loading tests. Laboratory findings include mildly elevated transaminases without signs of any impairment of synthetic or secretory function. If a liver biopsy is performed, both histological and biochemical methods show an increased glycogen content; however, enzymatic studies of all glycogenolytic enzymes are normal. Hyperami-noaciduria, hyperphosphaturia, hypercalciuria, renal tubular acidosis, mild tubular proteinuria, and polyuria are indicative of a generalized proximal tubular dysfunction. A hallmark to the diagnosis of FBS is the relatively severe glucosuria. The diagnosis of FBS is ultimately confirmed by the detection of homozygosity or compound heterozygosity for SLC2A2 mutations.40
 TREATMENT AND PROGNOSIS
TREATMENT AND PROGNOSIS
Only symptomatic treatment is available. Measures are directed toward improving glucose homeostasis and ameliorating the consequences of renal tubulopathy. FBS patients should receive a diet with adequate caloric intake to compensate for the renal glucose losses. Frequent feeds using slowly absorbed carbohydrates are recommended. Continuous carbohydrate supply by tube feeding oligosaccharide solutions during the night may be indicated. Uncooked cornstarch has a beneficial effect on metabolic control, particularly on growth.45
Regarding tubulopathy, water and electrolytes have to be replaced in appropriate amounts. Administration of alkali may be necessary to compensate for renal tubular acidosis. Hypophosphatemic rickets requires supplementation with phosphate and vitamin D preparations. With these measures, prognosis is fairly good, and some of the originally described pediatric patients have reached adulthood. The main subjective problems for these adult patients are short stature and orthopedic problems caused by hypophosphatemic rickets and osteomalacia. 
ARTERIAL TORTUOSITY SYNDROME
Deficiency of the facilitative glucose transporter GLUT10—encoded by SLC2A10, which is located on chromosome 20q13.1—has only recently been shown to be the cause of arterial tortuosity syndrome (OMIM 208050).46 The mechanism by which the defect of a glucose transporter causes elongation and tortuosity of all major arteries and changes in the connective tissue is not understood. 
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


