Disorders of Fatty Acid Oxidation
Stephen I. Goodman
Mitochondrial fatty acid oxidation provides the main source of energy for heart and skeletal muscle and, by generating acetyl-CoA for ketone-body production, provides energy for other tissues when the supply of glucose is limited. Fatty acids entering the cell are esterified with carnitine before being transported across the mitochondrial membrane and being beta-oxidized in the mitochondrial matrix as CoA esters (Fig. 150-1). Disorders that decrease β-oxidation by blocking cellular carnitine uptake or by impairing entry of fatty acids into mitochondria or β-oxidation limit energy production by heart and skeletal muscle at rest and lessen the ability of other tissues, including the brain, to cope with a low-glucose milieu.
Most patients with such disorders present before the age of 2 years, about 25% of them in the first week of life. Neonates may present with cardiac arrhythmias or sudden death, occasionally with facial dysmorphism and malformations, including renal cystic dysplasia. Symptoms in infancy and early childhood may relate to the liver or to cardiac or skeletal muscle, and thus include fasting- or stress-related hypoketotic hypoglycemia or Reye-like syndrome; conduction abnormalities; arrhythmias or dilated or hypertrophic cardiomyopathy; and muscle weakness or fasting- and/or exercise-induced rhabdomyolysis.
Diagnosis may be difficult even when the presentation is characteristic. Probably the most important single diagnostic test is analyzing acylcarnitine esters in serum or plasma by tandem mass spectroscopy (MS-MS), which identifies characteristic esters in many disorders, even when patients are symptom-free. Other tests that may be useful include analysis of urine organic acids and free and total carnitine in serum and urine, loading tests with medium- and long-chain fats, and enzyme assays in leukocytes or fibroblasts.
Treatment of acute encephalopathy associated with hypoketotic hypoglycemia is with 10% glucose and L-carnitine, given intravenously. Long-term therapy involves replenishing carnitine stores with L-carnitine and preventing hypoglycemia. This may be accomplished by providing a snack before bedtime, but continuous intragastric feeding may be required. Except for MCAD deficiency and the disorders that respond dramatically to carnitine (eg, carnitine uptake defect), the long-term prognosis for most of these conditions is guarded, with sudden death often occurring due to a conduction defect or an arrhythmia.1-4
CARNITINE UPTAKE DEFECT (PRIMARY CARNITINE DEFICIENCY)
This disorder is caused by a defect in the sodium-dependant high-affinity carnitine transporter in the plasma membrane, which ultimately limits β-oxidation by reducing entry of acyl-CoA esters into mitochondria (see Fig. 150-1). The defects in the kidney and gut cause very low levels of free carnitine in serum, and the disorder responds dramatically to L-carnitine. It is inherited as an autosomal recessive trait.5
 CLINICAL FEATURES
CLINICAL FEATURES
The condition may present in early infancy or in late childhood, usually with dilated cardiomyopathy or recurrent episodes of encephalopathy and hypoketotic hypoglycemia. Skeletal muscle involvement may be apparent as hypotonia or proximal limb weakness. Conduction defects and arrhythmias are rare. Sudden death is common, with autopsy showing fat in the heart, liver, renal tubules, and skeletal muscle.
 DIAGNOSIS
DIAGNOSIS
Diagnosis is based on finding extremely low levels of carnitine in serum and tissues; serum carnitine may be 1 μmol/L or undetectable (normal is 30–70). Organic acid and acylcarnitine analysis are usually normal. Carnitine transport is deficient in fibroblasts, and fetal diagnosis is possible based on the same assay on amniocytes. The gene encoding the carnitine transporter has been cloned and localized to chromosome 5 (5q31), and several disease-causing mutations have been described.
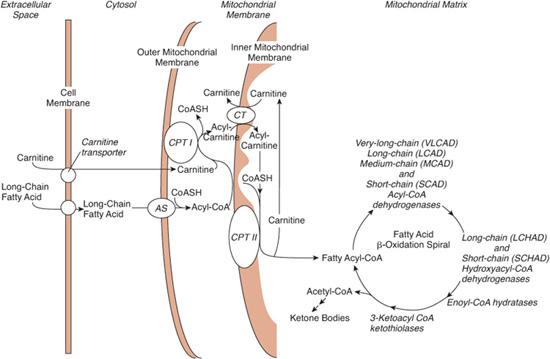
FIGURE 150-1. Transport and metabolism of fatty acids. To cross the mitochondrial membrane, long-chain fatty acids must be ligated to carnitine by carnitine palmitoyltransferase I (CPTI) and transferred by a translocase (CT). Carnitine palmitoyltransferase II (CPTII) releases the acyl group from carnitine into the mitochondrial matrix. Medium- and short-chain fatty acids can freely enter the mitochondria and do not require the carnitine system. Fatty acids are oxidized in a cycle that removes one acetyl-CoA moiety per turn. Dehydrogenases specific to very-long-, long-, medium-, and short-chain fatty acids catalyze the first reaction. As described in the text, defects have been found in many of the transporters and enzymes shown. Ketone bodies are formed in the liver from acetyl-CoA moieties. All defects of fatty acid oxidation are inherited as autosomal recessive diseases. AS, acyl-CoA synthetase; CT, carnitine-acylcarnitine translocase.
 TREATMENT
TREATMENT
The response to L-carnitine supplementation is dramatic and lifesaving; 100 to 200 mg/kg per day can be given intravenously in emergency situations, and then administered orally.
DEFECTS OF FATTY ACID ENTRY INTO MITOCHONDRIA
Short- and medium-chain fatty acids can enter mitochondria directly, but fatty acids longer than C12 must be esterified with coenzyme A and transported across the mitochondrial membrane by carnitine palmitoyltransferases (CPT) I and II, and carnitine-acylcarnitine translocase (see Fig. 150-1). CPT I in the outer membrane first transfers the acyl moieties from CoA to carnitine, the translocase moves the carnitine esters across the inner membrane, and CPT II on the matrix side of the inner membrane reconstitutes the CoA esters, which enter the β-oxidation spiral. Defects of CPT I, CPT II, and translocase are inherited as autosomal recessive traits.6-8
 CLINICAL FEATURES
CLINICAL FEATURES
CPT I deficiency usually presents in infancy with episodes of fasting-induced hypoketotic hypoglycemia. There are two distinct pheno-types of CPT II deficiency. The more common muscular form presents with exercise-induced muscle pain and rhabdomyolysis in adult life; only the rare hepatocardiomuscular form occurs in infants and children. Those who present as neonates die in 1 to 2 weeks with hepatomegaly, cardiomyopathy, and encephalopathy, and they often have renal cystic dysplasia. Those presenting later usually have hypoketotic hypoglycemia, cardiomyopathy, and muscle disease and are at risk for sudden death due to conduction defects or arrhythmias.
Translocase deficiency usually presents as life-threatening disease in the newborn period with hyperammonemia, conduction defects, arrhythmias, and evidence of skeletal muscle involvement, with high creatine phosphokinase. The ultimate prognosis of this condition is good if the child survives the neonatal period.
 DIAGNOSIS
DIAGNOSIS
The serum acylcarnitine profile is usually normal in carnitine palmitoyltransferase (CPT) I deficiency but may show elevated C16 esters in CPT II and translocase deficiency. There are no changes in amino acids (eg, high or low citrulline) to indicate a urea cycle defect as the cause of the hyperammonemia in translocase deficiency, and urine organic acids are normal or show only mild dicarboxylic aciduria. Free carnitine concentration in serum is 2 to 3 times normal in CPT I deficiency and is very low in CPT II and translocase deficiency.
All three enzymes can be assayed in fibro-blasts and leukocytes, and while prenatal diagnosis by enzyme assay on amniocytes should be possible in all of these disorders, it has been accomplished only in CPT II and translocase deficiency. In some instances, prenatal diagnosis of CPT II can also be made by ultra-sonography that shows enlarged echogenic kidneys. Disease-causing mutations have been identified in all the genes.
 TREATMENT
TREATMENT
Acute episodes of hypoketotic hypoglycemia should be treated with intravenous glucose-containing fluids, and treatment of hyperammonemia may require dialysis (see Fig. 145-4). Preventing fasting is simple in CPT I deficiency, but continuous intragastic feeding may be necessary in CPT II and translocase deficiency. Carnitine should be given when the serum carnitine level is low.
DEFECTS OF β-OXIDATION
Once in the mitochondrial matrix, acyl-CoA esters enter the β-oxidation spiral, where a series of four reactions remove two-carbon fragments of acetyl-CoA. Flavin adenine dinucleotide (FAD)-containing acyl-CoA dehydrogenases oxidize the acyl-CoA to 2,3-enoyl-CoA, which becomes hydrated to a 3-hydroxyacyl-CoA by hydratases. Oxidation to a 3-ketoacyl-CoA by nicotinamide adenine dinucleotide (NAD)-requiring hydroxyacyl-CoA dehydrogenases and removal of acetyl-CoA by 3-ketothiolases follow, and the acyl-CoA, now two carbons shorter, reenters the spiral.
All these reactions are catalyzed by enzymes with distinct (and often overlapping) chain-length specificities. For example, different FAD-containing acyl-CoA dehydrogenases act on very-long-chain (C12–24), long-chain (C6–20), medium-chain (C4–14), and short-chain (C4–6) acyl-CoAs. Similar specificities exist for the hydratases, hydroxyacyl-CoA dehydrogenases, and thiolases.
Inherited disorders in almost all these enzymes have been described. As a rule, defects in long-chain-specific enzymes block β-oxidation more completely and cause more severe clinical disease. Most of these conditions are rare, but very-long-chain acyl-CoA dehydrogenase (VLCAD) deficiency, medium-chain acyl-CoA dehydrogenase (MCAD) deficiency, and long-chain hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency are relatively common and therefore merit further discussion. All three are inherited as autosomal recessive traits. Short-chain acyl-CoA dehydrogenase deficiency is frequently suspected on neonatal screening on the basis of ethylmalonic and butyryl carnitine accumulation, but the molecular basis of the disease remains unclear, as many individuals remain asymptomatic without treatment.9
VERY-LONG-CHAIN ACYL-CoA DEHYDROGENASE (VLCAD) DEFICIENCY
 CLINICAL FEATURES
CLINICAL FEATURES
VLCAD deficiency can present in the newborn period with arrhythmias and sudden death, or with hepatic, cardiac, or muscle presentations later in infancy or childhood. The hepatic presentation is characterized by fasting-induced hypoketotic hypoglycemia, encephalopathy, and mild hepatomegaly, often with mild acidosis, hyperammonemia, and elevated liver transaminases. Some patients present with arrhythmias or dilated or hypertrophic cardiomyopathy in infancy or childhood, and a few patients present with exercise-induced muscle pain, rhabdomyolysis, elevated creatine kinase levels, and myoglobinuria. Most VLCAD-deficient patients present early with severe cardiomyopathy, and their outcome is poor.
 DIAGNOSIS
DIAGNOSIS
Analysis of serum acylcarnitines by tandem mass spectroscopy (MS-MS) usually shows elevations of saturated and unsaturated C14–18 esters, even between episodes. Urine organic acid analysis during acute illness often shows increased C6 (adipic), C8 (suberic), and C10 (sebacic) dicarboxylic acids. However, because very similar changes are seen in resolving ketosis and after ingesting medium-chain triglycerides, this will not raise suspicion of disease unless C12 and C14 dicarboxylic acids are also present. Serum concentration of free carnitine is usually low.
VLCAD deficiency can be demonstrated in fibroblasts or leukocytes, and enzyme assay in cultured amniocytes can be used for prenatal diagnosis. The gene encoding the enzyme has been cloned and localized to 17p13. Several disease-causing mutations have been described, and those that mainly affect enzyme function cause the most severe and early presenting clinical disease.
 TREATMENT
TREATMENT
Management involves avoiding fasting, maintaining high carbohydrate intake, and treating episodes of acute disease with glucose-containing intravenous fluids. Continuous intragastric feeding may be necessary. Oral carnitine may also be useful. Medium-chain triglycerides, whose oxidation does not involve VLCAD, can be administered to provide calories but should not be used until the diagnosis of VLCAD deficiency is certain.10-12
MEDIUM-CHAIN ACYL-CoA DEHYDROGENASE (MCAD) DEFICIENCY
 CLINICAL FEATURES
CLINICAL FEATURES
MCAD deficiency usually presents during the first 2 years of life with episodes of fasting-induced vomiting, lethargy progressing to coma and seizures, hypoketotic hypoglycemia, and hepatomegaly, often with mild hyperammonemia. Misdiagnosis as Reye syndrome or, because the initial episode is fatal in about 25% of patients, sudden infant death syndrome is common. Levels of uric acid, liver transaminases, and creatine kinase are often elevated during the acute episode, and liver biopsy shows microvesicular steatosis. Autopsy shows fatty infiltration of the liver, renal tubules, and heart and skeletal muscle. A few enzyme-deficient individuals do not develop symptoms.
 DIAGNOSIS
DIAGNOSIS
Analysis of serum acylcarnitines by MS-MS shows elevations of C8, C8:1, and C10:1 esters, even between episodes. Urine organic acids during acute illness often show increased C6 (adipic), C8 (suberic), and C10 (sebacic) dicarboxylic acids, together with hexanoylglycine, suberylglycine, and phenylpropionylglycine. The dicarboxylic aciduria also occurs during resolving physiological ketosis and after ingestion of medium-chain triglycerides. It will not be perceived as abnormal unless the glycine esters, or unsaturated or longer-chain dicarboxylic acids, are also present. Free-carnitine level in serum is usually low.
The enzyme defect can be demonstrated in several tissues, including fibroblasts and leukocytes, but molecular diagnosis is often easier. The gene encoding MCAD has been localized to 1p31. Several disease-causing mutations have been identified, but the K304E mutation, which changes the lysine residue at position 304 of the mature enzyme to glutamic acid, accounts for 90 percent of mutant alleles found in whites. Ninety-nine percent of affected whites have at least one copy of this mutation (81% are homozygous, and 18% are heterozygous), and analysis for this one mutation will confirm the diagnosis in many patients.
 TREATMENT
TREATMENT
Acute episodes should be treated with intravenous glucose and bicarbonate. Long-term treatment consists of avoiding fasting, usually by providing carbohydrate snacks at bedtime. Oral carnitine (100 mg/kg per day) is useful, both to replenish depleted stores and to augment excretion of toxic intermediates as carnitine esters. Developmental delay, behavioral problems, and other chronic CNS problems are not uncommon outcomes of the initial episode, but if such damage does not occur, prognosis is quite good. Newborn screening for MCAD deficiency has been advocated and is based on the responsiveness to treatment and the frequency of death and other sequelae following the initial episode (see Chapter 134).11
LONG-CHAIN 3-HYDROXYACYL-CoA DEHYDROGENASE (LCHAD) DEFICIENCY
NAD-dependent 3-hydroxyacyl-CoA dehydrogenases catalyze oxidation of 3-hydroxyacyl-CoA esters to their 3-keto analogues. LCHAD, the long-chain-specific enzyme, acts on acyl groups longer than C8 and exists in mitochondria as part of a protein with three activities. This mitochondrial trifunctional protein (MTP) is an α4β4 octamer, with the α subunit carrying LCHAD and long-chain enoyl-CoA hydratase, and the β subunit carrying long-chain β-ketothiolase. LCHAD deficiency can exist alone or with deficiency of the other two enzymes.
 CLINICAL FEATURES
CLINICAL FEATURES
Patients with isolated LCHAD deficiency often present in infancy with fasting-induced hypoketotic hypoglycemia; however, patients may also present with neonatal cardiomyopathy or, much later, with exercise-induced rhabdomyolysis. History often reveals a pregnancy complicated by acute fatty liver or HELLP (hemolysis, elevated liver enzymes, and low platelets). Unlike most other fatty acid oxidation disorders, severe and progressive cholestatic liver disease is common, and many patients develop retinopathy with hypopigmentation or focal pigmentary aggregations. Most patients die in early childhood. The presentation of individuals with MTP deficiency is similar to that of isolated LCHAD deficiency, but it is usually earlier and more severe.
 DIAGNOSIS
DIAGNOSIS
Acylcarnitine analysis by MS-MS is usually diagnostic and shows elevated saturated and unsaturated C16 and C18 hydroxyacylcarnitines. Organic acid analysis often shows elevated C6–14 3-hydroxydicarboxylic acids, but the same abnormalities are seen in patients with respiratory-chain defects and glycogenoses and are not specific. The enzyme defect can be demonstrated in fibroblasts and leukocytes and, for prenatal diagnosis, in amniocytes. The gene encoding the α subunit has been cloned and localized to 2p24.1-23.3. The E510Q mutation, which changes the glutamic acid residue at position 510 to glutamine, accounts for nearly 90% of mutant alleles in Europeans.
 TREATMENT
TREATMENT
Therapy is much the same as that of VLCAD and MCAD deficiency. As in VLCAD deficiency, it may be necessary to completely eliminate fasting by continuous intragastric feeding, and medium-chain triglycerides may be used to provide calories. Carnitine is probably useful to replenish depleted stores. Oral supplements of docosahexaenoic acid, a polyunsaturated C20 acid, may be useful in reversing retinopathy.13,14
GLUTARIC ACIDEMIA TYPE II
Electrons from the acyl-CoA dehydrogenases involved in fatty acid and amino acid oxidation are transferred from their FAD coenzymes into the respiratory chain via redox centers in electron transfer flavoprotein (ETF) and ETF: ubiquinone oxidoreductase (ETF:QO). Recessively inherited defects in ETF and ETF:QO cause glutaric acidemia type II (GA2).
 CLINICAL FEATURES
CLINICAL FEATURES
GA2 may present in the neonatal period with severe hypoglycemia, metabolic acidosis, hyperammonemia, and the odor of sweaty feet typical of isovaleric acidemia, often with cardiomyopathy, facial dysmorphism, and severe renal cystic dysplasia. Most such patients die within the first days or weeks of life, often of conduction defects or arrhythmias. Fatty infiltration of the liver, renal tubules, and heart and skeletal muscle are consistent autopsy findings. Milder disease, sometimes called ethylmalonic adipic aciduria, may present with episodic hypoketotic hypoglycemia and hepatomegaly in childhood, or simply as hypoglycemia in adult life.
 DIAGNOSIS
DIAGNOSIS
Acylcarnitine analysis by MS-MS shows glutarylcarnitine, isovalerylcarnitine, and straight-chain esters of chain lengths C4, C8, C10, C10:1, and C12. Urine organic acids show increased ethylmalonic, glutaric, 2-hydroxyglutaric, and 3-hydroxyisovaleric acids, together with C6, C8, and C10 dicarboxylic acids and isovalerylglycine. Serum carnitine concentration is usually low. Elevated serum sarcosine on amino acid analysis is common in patients with mild disease.
Assays on fibroblasts show that some GA2 patients are deficient in ETF and that others are deficient in ETF:QO. The genes encoding ETF:QO and the α and β subunits of ETF have been cloned and mapped, and disease-causing mutations in all three genes have been identified in different patients.
 TREATMENT
TREATMENT
Patients with complete defects die during the first weeks of life, usually of conduction defects or arrhythmias; but those with incomplete defects can survive well into adult life. As in other fatty acid oxidation disorders, treatment relies on the avoidance of fasting, sometimes with continuous intragastric feeding, and carnitine to replenish lost stores. Medium-chain triglycerides cannot be oxidized in this condition, because all of the acyl-CoA dehydrogenases are deficient.15,16
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


