Disorders of Cholesterol Synthesis
Peter T. Clayton
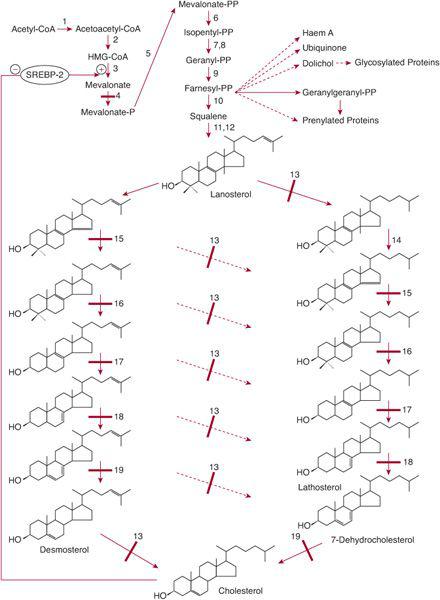
The isoprenoid synthesis pathway (Fig. 164-1) leads to the synthesis of cholesterol, haem A, ubiquinone, dolichol, farnesyl-pyrophosphate, and geranylgeranyl-pyrophosphate. 
When cholesterol levels are low, transcription of the HMG-CoA reductase gene is upregulated. 
Cholesterol is an important constituent of cell membranes and has important interactions with signaling proteins that are involved in embryogenesis and development (eg, the hedgehog proteins). Disorders of the pathway from lanosterol to cholesterol lead to dysmorphic features and to malformations of internal organs. In all but one of these disorders, psychomotor delay is prominent. 
MEVALONATE KINASE DEFICIENCY (MEVALONIC ACIDURIA [MA] AND HYPER-IGD AND PERIODIC FEVER SYNDROME [HIDS])
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Mevalonic aciduria (MA) is the severe form of mevalonate kinase deficiency; the hyper-IgD periodic fever syndrome (HIDS) is more benign. Both present in infancy with episodes of pyrexia for 3 to 5 days every 4 to 6 weeks; symptoms include diarrhea and vomiting, abdominal pain, cervical lymphadenopathy, hepatosplenomegaly, arthralgia, and a rash.3,4 Children with MA have additional problems. Dysmorphic features include microcephaly, large fontanels, triangular face, low-set ears, down-slanting palpebral fissures, and cataracts. Neurological manifestations include hypotonia, developmental delay, nystagmus, and progressive ataxia. Babies with MA may die in infancy. 
 METABOLIC DERANGEMENT
METABOLIC DERANGEMENT
MA and HIDS are both caused by a deficiency of mevalonate kinase. In MA, the activity of mevalonate kinase in white cells or cultured skin fibroblasts is undetectable, whereas in HIDS it is 2% to 8% that of controls.5
 GENETICS
GENETICS
MA (OMIM 610377) and hyper-IgD periodic fever syndrome (HIDS) (607622) are autosomal recessive conditions caused by mutations in the MK gene, which encodes mevalonate kinase and is located on chromosome 12q24.5 The majority of patients with HIDS are compound heterozygotes for a V377I mutation (found only in patients with HIDS), and a second mutation that can be one of the mutations found in mevalonic aciduria. Prenatal diagnosis can be undertaken by measuring MK activity or mutation detection in chorionic villus, cultured villus cells, and amniotic fluid cells. 
 DIAGNOSTIC TESTS
DIAGNOSTIC TESTS
Measurement of urinary mevalonic acid will readily detect patients with mevalonic aciduria but may not detect all patients with HIDS.6 Most HIDS patients have elevated plasma IgD and/or IgA. The best tests are measurement of MK activity in white blood cells/cultured skin fibroblasts and sequencing of the MVK gene. 
 TREATMENT AND PROGNOSIS
TREATMENT AND PROGNOSIS
Mevalonic aciduria can be fatal, but the prognosis for HIDS is good with symptoms subsiding with age.4 No treatment has been shown to be effective in all patients.7-9 One patient has recently been successfully treated by allogenic bone marrow transplant.10
SMITH-LEMLI-OPITZ SYNDROME (SLOS; 7-DEHYDROCHOLESTEROL REDUCTASE DEFICIENCY)
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
The symptoms and signs of Smith-Lemli-Opitz syndrome (SLOS) vary enormously in severity, from intrauterine death in the first or second trimester to subtle abnormalities leading to diagnosis of the disorder in an adult whose educational attainment has been normal. Typical features of the disorder include intrauterine growth retardation with low maternal estriol levels; sometimes abnormalities are detected on ultrasound, such as increased nuchal translucency, polydactyly, and facial dysmorphism. At birth, dysmorphic features may be obvious11,12: microcephaly; ptosis; anteverted nares; long philtrum; low-set, posteriorly rotated ears; micrognathia; cleft or high-arched palate; syndactyly of toes 2, 3; postaxial polydactyly; and short, proximally placed thumbs. Boys may have hypospadias and cryptorchidism or even ambiguous or female genitalia. Hypotonia is common. Serious congenital malformations include airway problems, congenital heart disease, pyloric stenosis, and Hirschsprung disease. Feeding problems and failure to thrive are common. However, the most common problem is developmental delay often associated with communication difficulties (akin to autism) and behavior problems, including aggression and sleep disruption. 
 METABOLIC DERANGEMENT
METABOLIC DERANGEMENT
SLOS is caused by a deficiency of 7-dehydrocholesterol (7-DHC) reductase,13 which catalyzes the last (or penultimate) step in cholesterol synthesis (Fig. 164-1). Tissues of patients have an increased concentration of 7-DHC and 8-dehydrocholesterol, or 8-DHC. 
 GENETICS
GENETICS
SLOS (OMIM 270400) is an autosomal recessive disorder caused by mutations in the DHCR7 gene encoding 7-DHC reductase on chromosome 11q13.14 Prenatal diagnosis can be made by analyzing maternal urine for dehydro-estriol and dehydro-pregnanetriol15 by analysis of sterols in chorionic villus or amniotic fluid16,17 or by measurement of 7-DHC reductase activity in villus/cultured villus cells.18 DNA analysis is the most reliable technique.19
 DIAGNOSTIC TESTS
DIAGNOSTIC TESTS
Analysis of plasma sterols shows elevated concentrations of 7-DHC and 8-DHC. The activity of 7-DHC reductase can be measured in white cells or fibroblasts. Mutations in the DHCR7 gene can be detected by sequencing. 
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


