Disorders of Bone Formation
Allen W. Root
NORMAL SKELETON: CARTILAGE AND BONE
The skeleton comprises cartilage and bone and is a support for the insertion of muscles and tendons enabling movement, a shield for soft tissue organs, a site for storage of bone marrow, and a reserve pool of calcium, phosphate, and other ions. There are flat (eg, cranium, scapula, pelvis) and long (eg, humerus, femur, phalanges) bones. The outer surface of bone is enclosed in periosteum (a fibrous network of osteoblasts that synthesizes peripheral compact bone); endosteum lines its inner surface. The central portion of long bones is a hollow shaft of dense cortical bone termed the diaphysis; at both ends of many long bones are the metaphysis (composed of cortical and cancellous or trabecular bone), cartilaginous growth plate, and epiphysis.
Osteoblasts are bone-forming cells derived from stromal mesenchymal stem cells that secrete collagen types I and III and noncollagenous proteins that together form osteoid or bone matrix into which calcium and phosphate are deposited. Osteoclasts are bone resorbing cells that are derived from hematopoietic precursor cells. Bone modeling is the process by which the shape and size of a bone is determined and occurs only during intrauterine development and postnatal growth. Bone remodeling is a process in which formed bone is replaced by new bone and takes place throughout life.
Eighty percent of the skeleton is composed of cortical bone present in the cranium, scapula, mandible, ilium, and shafts of the long bones. Cancellous or trabecular bone is found in vertebrae, skull base, pelvis, and ends of the long bones. Since only 15% to 25% of trabecular bone is calcified, it has a large surface area and a high turnover rate and is particularly susceptible to disorders that affect bone mineralization. Intramembranous ossification is initiated by local condensation of mesenchymal stem cells that differentiate directly into osteoblasts. When intracellular levels of β-catenin are low, the mesenchymal stem cell evolves into a chondroblast. Longitudinal growth of bone is the result of chondrocyte proliferation in orderly columns within the cartilaginous growth plate (see eFig. 543.1  ). Resting or reserve cells at the most distal aspect of the growth plate differentiate into proliferating chondrocytes under stimulation by bone morphogenetic protein-6 and growth hormone. Locally synthesized insulin-like growth factor I(generated in response to growth hormone, thyroid and sex hormones) stimulates division of these chondrocytes and increase in the length of the cartilage growth plate and of the long bone. Endochondral bone develops as invading blood vessels bring with them chondroclasts, osteoclasts, and osteoblasts; chondroclasts reabsorb cartilage matrix and dying chondrocytes. Primary spongiosa of bone is formed and then replaced by mature bone. In addition to collagen type I, osteoblasts secrete many noncollagenous proteins (eg, bone-specific alkaline phosphatase, osteocalcin) into a matrix into which calcium and phosphate are deposited as hydroxyapatite. Osteoblasts have a life span of 3 months.
). Resting or reserve cells at the most distal aspect of the growth plate differentiate into proliferating chondrocytes under stimulation by bone morphogenetic protein-6 and growth hormone. Locally synthesized insulin-like growth factor I(generated in response to growth hormone, thyroid and sex hormones) stimulates division of these chondrocytes and increase in the length of the cartilage growth plate and of the long bone. Endochondral bone develops as invading blood vessels bring with them chondroclasts, osteoclasts, and osteoblasts; chondroclasts reabsorb cartilage matrix and dying chondrocytes. Primary spongiosa of bone is formed and then replaced by mature bone. In addition to collagen type I, osteoblasts secrete many noncollagenous proteins (eg, bone-specific alkaline phosphatase, osteocalcin) into a matrix into which calcium and phosphate are deposited as hydroxyapatite. Osteoblasts have a life span of 3 months.
Stromal cells and osteoblasts also control the differentiation, maturation, and function of bone-resorbing osteoclasts. Expressed on the cell membrane of osteoblasts and stromal cells is the receptor-activator for nuclear factor κB-ligand (RANKL), a protein that stimulates osteoclasto-genesis through interaction with RANK expressed on the cell membrane of osteoclast progenitor cells. Stromal cells and osteoblasts also synthesize and secrete osteoprotegerin—a decoy protein than binds RANKL, thereby inhibiting osteoclastogenesis. Parathyroid hormone, calcitriol, and cytokines (eg, interleukins, tumor necrosis factor-α) stimulate production of both RANKL and osteoprotegerin. After binding of RANKL to RANK, the progenitor cell differentiates and becomes a functional osteoclast. Under the direction of RANKL, the osteoclast produces specific proteins such as tartrate-resistant acid phosphatase, cathepsin K, β3-integrin, and the calcitonin receptor. The production of osteoprotegerin is also stimulated by BMP and estrogen; synthesis is inhibited by glucocorticoids and prostaglandins (see eFig. 543.2  ).
).
Osteoclasts are multinucleated giant cells that attach to bone and form a tightly sealed space (lacuna) between the cell’s basal ruffled membrane and the bone’s outer surface (see eFig. 543.3  ). Osteoclasts reabsorb bone by pumping hydrogen and chloride ions into the lacunae that dissolve hydroxyapatite and enzymes (eg, cathepsins, matrix metalloproteinases) that degrade the protein matrix of bone. Acidification of the subosteoclastic space requires the action of carbonic anhydrase II and proton and chloride channels. The dissolved minerals and degraded proteins are absorbed by the osteoclast and extruded into the circulation. When local bone resorption is complete, osteoclasts leave the site.
). Osteoclasts reabsorb bone by pumping hydrogen and chloride ions into the lacunae that dissolve hydroxyapatite and enzymes (eg, cathepsins, matrix metalloproteinases) that degrade the protein matrix of bone. Acidification of the subosteoclastic space requires the action of carbonic anhydrase II and proton and chloride channels. The dissolved minerals and degraded proteins are absorbed by the osteoclast and extruded into the circulation. When local bone resorption is complete, osteoclasts leave the site.
The initial process of bone modeling (ie, embryonic bone formation) is accomplished by the action of osteoblasts and osteoclasts acting independently of one another and is not dependent upon prior bone resorption. Bone remodeling is a continuous process in which old bone is replaced by new bone. Bone remodeling occurs in both growing and mature bone either at random locations or at sites of mechanical stress. 
Serum measurements of the C-terminal extension of procollagen type I, osteocalcin, and bone-specific alkaline phosphatase provide information about osteoblast function and bone formation. After collagen type I is degraded by osteoclasts, PYR, DPD, and the N-telopeptides (NTx, the amino terminal telopeptide of type I collagen) and C-telopeptides (CTx; carboxyl terminal telopeptide of type I collagen) are released; their serum and urine levels serve as indices of bone resorption. Reference levels of markers of bone formation and bone resorption in neonates, infants, children, and adolescents are recorded in eTables 543.1 and 543.2  .
.
The average total body bone mineral content (BMC) is 2800 g in the adult male and 2200 g in the adult female, the majority of which (60%) is laid down during puberty. It is currently recommended that during adolescence, 1300 to 1600 mg of elemental calcium be consumed daily in order to reach maximal adult bone mineral mass. Sixty percent to 80% of peak adult bone mass is determined by familial genetic factors as evidenced by the concordance between bone mineral density (BMD) levels in mothers and daughters, identical and fraternal twins, and siblings. Whole body, lumbar spine, total hip, and femoral neck BMD and bone mineral apparent density are greater in black youth than in white, Asian, or Hispanic youth. Radial, femoral neck, and vertebral BMDs correlate with sex, age, height, weight, body mass index, pubertal and postpubertal hormonal status, calcium intake, and exercise in children, adolescents, and adults. Bone mass is also closely related to weight. Obese subjects have greater bone mass than do slim individuals. However, in obese subjects BMC and BMD are related primarily to lean body mass (ie, muscle) rather than to fat mass or volume. 
Limitation of movement, physical activity, and weight bearing adversely affect bone growth and strength. Thirty minutes of weight-bearing exercise 3 times weekly increases femoral neck and lumbar spine bone mineral content in pre-pubertal children. High-impact activities such as ballet, tennis, gymnastics, and soccer substantially increase periosteal bone formation, cortical thickness, and mass of weight-bearing bones in children and adolescents. When weight bearing is decreased by immobilization or decreased gravity (eg, space flight), bone loss (disuse osteoporosis) ensues because of decrease in mechanical forces.
EFFECTS OF HORMONES AND GROWTH FACTORS ON THE SKELETON
Chondrocyte proliferation, maturation, and function are regulated by systemic hormones and growth factors (growth hormone, insulin-like growth factor-I [IGF-I], parathyroid hormone, leptin, thyroid and sex hormones, glucocorticoids). Growth hormone increases chondrocyte differentiation and proliferation in the reserve zone and enhances local expression of IGF1. Fetal growth is dependant on both IGF-I and IGF-II and the IGF-I receptor. Growth hormone promotes osteoblast differentiation and proliferation and the synthesis of bone-specific alkaline phosphatase, collagen type I, and IGF-I. Administration of growth hormone increases serum and urine values of markers of bone formation and resorption (osteocalcin, bone-specific alkaline phosphatase, pyridinoline, deoxypyridinoline, N-telopeptide-NTx). Bone mineral content and bone mineral density are decreased in growth hormone-deficient patients and increase during administration of growth hormone. In patients with isolated deficiency of IGF-I due to deletion of IGF1, bone size is small, and administration of IGF-I increases bone mineral content and bone mineral density. 
Thyroid hormones increase osteoclast numbers and bone resorption and remodeling and in excess lead to net bone loss. Both estrogens and androgens stimulate proliferation and maturation of chondrocytes and long bone growth. Androgens and estrogens are essential for accretion of bone mineral. Although bone mineral accrual is greater in males than females, it is the effect of estrogen that predominates in this process as demonstrated by the observations that adult androgen-sufficient males with impaired aromatase activity or estrogen receptor function are markedly osteopenic and that estrogens but not testosterone can increase bone mass in males with aromatase deficiency. Glucocorticoids suppress osteoblast formation and accelerate their death rate, thus impairing bone formation. They depress osteoclast formation but increase their life span. Taken together, these actions lead to osteopenia and osteoporosis.
Adipocytes synthesize adipokines that affect bone development.
ASSESSMENT OF BONE MASS AND STRENGTH
Histomorphometric analysis of undecalcified transiliac bone biopsies containing trabecular and cortical bone permits examination and quantitation of the processes and rates of bone formation, resorption, modeling, and remodeling. Bone size, mass, and strength can be assessed quantitatively by noninvasive methods such as standard bone x-rays (of limited value), dual-energy x-ray absorptiometry (DEXA), axial and peripheral quantitative computed tomography, metacarpal radiogrammetry, quantitative ultra-sonography, quantitative magnetic resonance, and magnetic resonance microscopy.
Because of its low radiation dosage (∼ 5 μSv), ease of use, applicability for infants, rapidity (4 minutes for total body scan), precision, and reproducibility, DEXA is the procedure that has been most frequently employed for assessment of axial (head, spine) and appendicular (limbs) bone mass and bone area as well as body composition (lean and fat masses) in children and adults.  Software developed specifically for infants and children must be used to analyze DEXA data in these populations. Since DEXA does not use the depth or width of a bone in its computations, it underestimates bone mineral density (BMD) in children with small bones and overestimates it in subjects with large bones. Since bone size or volume increases with growth and maturation in children and adolescents, the larger a bone, the greater the recorded areal BMD, although the volumetric BMD need not change appreciably. The bone mineral apparent density (BMAD; in g/cm3) has been calculated from DEXA data to address this problem. BMAD approximates volumetric BMD. Whole body BMC and BMD, lumbar spine BMC and BMD, and femoral BMD are the most commonly measured DEXA indices of bone mass in childhood. DEXA data in children are reported as a z score, the number of standard deviations about the mean of gender and age-matched peers to which the observed figure corresponds. (The DEXA t score is the number of standard deviations about the mean of peak bone mass recorded in healthy young adults aged 20 to 29 years. The t score is commonly reported in adults but should not be employed in children and adolescents.) In growing infants, children, and adolescents, whole or total body BMC is a reasonably accurate and reliable DEXA measurement as compared to regional measurements.
Software developed specifically for infants and children must be used to analyze DEXA data in these populations. Since DEXA does not use the depth or width of a bone in its computations, it underestimates bone mineral density (BMD) in children with small bones and overestimates it in subjects with large bones. Since bone size or volume increases with growth and maturation in children and adolescents, the larger a bone, the greater the recorded areal BMD, although the volumetric BMD need not change appreciably. The bone mineral apparent density (BMAD; in g/cm3) has been calculated from DEXA data to address this problem. BMAD approximates volumetric BMD. Whole body BMC and BMD, lumbar spine BMC and BMD, and femoral BMD are the most commonly measured DEXA indices of bone mass in childhood. DEXA data in children are reported as a z score, the number of standard deviations about the mean of gender and age-matched peers to which the observed figure corresponds. (The DEXA t score is the number of standard deviations about the mean of peak bone mass recorded in healthy young adults aged 20 to 29 years. The t score is commonly reported in adults but should not be employed in children and adolescents.) In growing infants, children, and adolescents, whole or total body BMC is a reasonably accurate and reliable DEXA measurement as compared to regional measurements.
In adults, osteopenia and osteoporosis refer to degrees of subnormal bone mass; osteoporosis is present when the site specific (lumbar spine, hip) bone mineral density (BMD) is –2.5 or more standard deviations below the gender-specific mean peak adult value (t score); osteopenia is present when the BMD lies between –1.1 and –2.4 standard deviations below the mean peak adult value.1 BMD is normal when it falls between +1 above and –1 standard deviations below the mean peak adult value. Bone mass is high when it is exceeds +2 standard deviations above the mean peak gender-specific adult value. Abnormalities of bone mass in children are usually designated as low or high (below –2 or above +2 standard deviations or z score for gender, respectively) in relationship to chronologic age, although height and stage of sexual maturation must also be considered when analyzing bone mass data.2,3 Most reference data for DEXA measurements of bone mass are related to age and not to body size. A short child may have a DEXA measurement that is below normal for age but reasonable for stature; therefore, assessment of DEXA data in relationship to height (and/or to bone age and/or stage of pubertal maturation) is important.
Quantitative ultrasound (QUS) estimates bone strength by measuring the speed of a longitudinal sound (SOS) wave as it moves along a bone. QUS does not use radiation and is low in cost, and the equipment is portable. There is only marginal correlation between volumetric BMD determined by peripheral quantitative computed tomography (QCT) and SOS measurements in children and adolescents. Nevertheless, QUS may complement radio-graphic methods of bone mineralization assessment. The method used for assessment of bone mineralization (DEXA, QCT, QUS) and the specific instrument, analytic software, and the ethnic mix of the reference population should be included in the interpretive report.4 Reference DEXA measurements are found in eTable 543.3 and eTable 543.4  .
.
DISORDERS WITH LOW BONE MASS DUE TO IMPAIRED BONE MINERALIZATION
 RICKETS AND OSTEOMALACIA
RICKETS AND OSTEOMALACIA
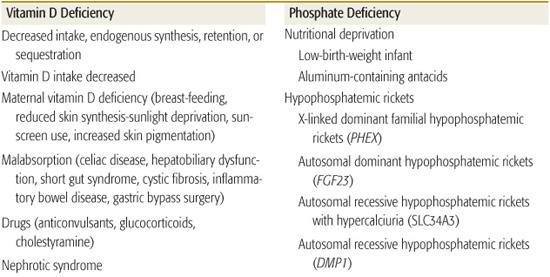
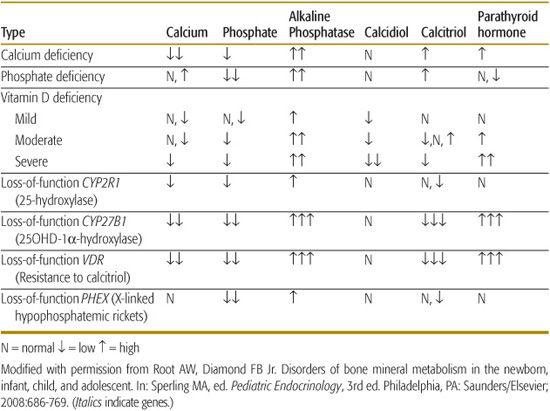
Rickets in children and osteomalacia in adults are due to decreased mineralization of bone matrix (Table 543-1).5,6 Rickets may be classified as calciopenic (due to deprivation of vitamin D or calcium) or phosphopenic (usually due to renal phosphate wasting). Typical patterns of laboratory values observed in patients with rickets due to various causes are shown in Table 543-2. Bone mineralization may also be impaired by abnormalities of alkaline phosphatase generation or by drugs that inhibit mineralization.7 Subnormal mineralization of endochondral bone decreases its strength; the ends of the weight-bearing long bones then deform.
Table 543-1. Disorders with Low Bone Mass Due to Impaired Bone Mineralization: Rickets

Clinical Features
Newborns of severely vitamin D–deficient mothers may have fractures and hypocalcemia. In preambulatory infants with rickets, bowing of the forearms, craniotabes (reversible compression of the skull’s outer table), frontal bossing, and delayed closure of the cranial fontanelles are observed. In children, genu varum or valgum (bowed legs or knock knees or a “windswept” deformity involving both legs), flaring of the metaphyses of the radius and ulna with enlarged wrists, prominence of the costochondral junctions (ricketic rosary), and indentation of the lower anterior thoracic wall (Harrison groove) are often present. Delayed eruption of teeth with hypo-plastic enamel, short stature, and suboptimal weight gain, hypotonia and delay in walking, anorexia, and increased susceptibility to infection are also observed in vitamin D-deprived children.8 Rickets is characterized radiographically by cupping, splaying, and fraying of the metaphyses of the long bones, bowing of the long bones, cortical narrowing, stress fracture lines, and diffuse demineralization.
 PREMATURITY/LOW BIRTH WEIGHT
PREMATURITY/LOW BIRTH WEIGHT
Since approximately 80% of total bone calcium is accrued in the last trimester of pregnancy and because low-birth-weight (LBW) (< 1500 g) and very LBW (< 1000 g) infants are unable to maintain the in utero rates of synthesis of bone matrix and mineralization from the nutrients absorbed from the gastrointestinal tract or administered by parenteral nutrition, they are at great risk for development of low bone mass (osteopenia of prematurity) and/or rickets.9-11 An immobilized premature or LBW male offspring of a multiparous mother who develops bronchopulmonary dysplasia and who is receiving glucocorticoids, theophylline, or furosemide is at particular risk for development of low bone mass.12,13 The LBW neonate is best managed preventively by the daily feeding of as much of the required amounts of calcium (140–160 mg/100 kcal of formula per day), phosphate (95–110 mg/100 kcal), vitamin D (400 U/day), protein, carbohydrates, and lipids as possible. Either breast milk fortified with calcium and phosphate or prepared formulas designed for the feeding of LBW neonates may be used. If parenteral administration of nutrients is necessary, the maximum safe amounts of calcium and phosphate should be infused. Frequent determinations of serum levels of calcium, phosphate, creatinine, and alkaline phosphatase and urinary excretion of calcium, phosphate, and creatinine are necessary to prevent hypocalcemia, hypercalcemia, hypercalciuria, and nephrocalcinosis. Although bone mass may remain low in the prematurely born infant through infancy and childhood, it eventually normalizes.14
 VITAMIN D DEFICIENCY OR INSUFFICIENCY
VITAMIN D DEFICIENCY OR INSUFFICIENCY
Vitamin D deficiency has reemerged as a common disorder of infants, children, and adolescents.15-18 Although grossly evident vitamin D–deficient rickets develops predominantly in dark-skinned infants and children who ingest a diet low in vitamin D without supplemental vitamin D and who have limited exposure to sunlight, subtle forms of vitamin D insufficiency are found in many North American and British adolescents. Approximately 40% of ostensibly healthy urban adolescents in the northeastern United States have serum concentrations of 25-hydroxyvitamin D (25OHD) less than 20 ng/mL, which is attributable to limited intake of milk and multivitamins, increasing consumption of phosphate-containing soft drinks, and increased adiposity, as fat is a repository for vitamin D. Vitamin D deficiency may also occur because of disorders of intestinal absorption (eg, celiac disease, biliary obstruction, gastric resection or by-pass surgery, pancreatic exocrine insufficiency) or by increased degradation to water-soluble forms increasing urinary loss, as seen with hepatic enzyme induction from anticonvulsant drugs such as phenytoin. In patients with vitamin D–deficient rickets, serum total calcium values are low-normal or low, phosphate levels are low, and alkaline phosphatase activity and parathyroid hormone concentrations are elevated; serum levels of 25-hydroxyvitamin D are low, and calcitriol values are variable (Table 543-1).
Vitamin D deficiency is most effectively managed by prevention. The breast-feeding mother should ingest 1000 IU of vitamin D daily, and breast-feeding infants should receive 400 IU of vitamin D daily. Infants and children who are not receiving adequate amounts of vitamin D in prepared formulas or in their diet and who have suboptimal exposure to sunlight should also receive 400 IU/day of vitamin D.19 Vitamin D supplementation (400–1000 IU/day) is appropriate throughout life.
Table 543-2. Laboratory Data in Rickets of Different Causes
The child or adolescent with vitamin D deficiency may be treated by oral administration of vitamin D 1000 to 10,000 IU daily for 8 to 12 weeks, or 50,000 IU weekly for 8 weeks, or a single oral dose of 150,000 to 600,000 IU, depending on the patient’s age and clinical status. When beginning treatment with vitamin D, elemental calcium (30–75 mg/kg/day in divided doses) should also be administered in order to prevent hypocalcemia accompanying rapid bone remineralization. The effect of therapy on the rachitic child should be monitored by serial measurements of urine and serum calcium levels and alkaline phosphatase values whose levels decline as the rachitic lesions heal.
 DEFECTS IN VITAMIN D SYNTHESIS AND FUNCTION
DEFECTS IN VITAMIN D SYNTHESIS AND FUNCTION
These disorders are rare causes of rickets. Homozygous inactivating mutations in the gene encoding 25-hydroxylase—CYP2R1—prevents synthesis of 25-hydroxyvitamin D (25OHD), leading to rickets, hypocalcemia, hypophosphatemia, increased alkaline phosphatase activity, and low serum levels of 25OHD.20 These patients respond to treatment with calcidiol. Loss of renal tubular 25OHD-1α hydroxylase activity due to inactivating mutations in CYP27B1 prevents synthesis of calcitriol, leading to vitamin D–dependent rickets type 1.21 Manifestations of this autosomal recessive disease in infancy include bowing of the forearms and of the legs, growth retardation, weakness, and hypocalcemic seizures. Laboratory findings include hypocalcemia, hypophosphatemia, hyperphosphatasemia, elevated serum parathyroid hormone levels, and normal concentrations of 25OHD but very low levels of calcitriol that do not increase after administration of vitamin D or calcidiol. The diagnosis of this disorder is confirmed by identification of the mutation in CYP27B1. Physiologic doses of calcitriol (10–20 ng/kg/day) reverse the clinical, biochemical, and radiographic abnormalities of vitamin D–dependent rickets type 1. Vitamin D–resistant rickets is an autosomal recessive disorder due to inactivating homozygous or compound heterozygous mutations of VDR, the gene encoding the vitamin D receptor. The clinical and biochemical findings in these infants are similar to those who lack 25OHD-1α hydroxylase activity, except the vitamin D–resistant patients often have alopecia (vitamin D is essential for growth of epithelial and outer root-sheath cells of the hair follicle) and serum concentrations of calcitriol are markedly elevated. High doses of calcitriol (1 to 6 μg/kg/day) and supplemental calcium (1–3 g of elemental calcium daily) increase serum calcium concentrations and heal rickets in some patients.22 Low calcium intake (< 200 mg/day) or diminished intestinal absorption of dietary calcium bound by fiber and phytate in cereals can lead to rickets in infants and children with normal intake of phosphate and serum levels of 25OHD.23 A child with calcium-deficiency rickets is effectively treated by intake of 1000 mg of elemental calcium daily for 6 months with provision of normal amounts of vitamin D.
 PHOSPHATE DEFICIENCY
PHOSPHATE DEFICIENCY
Hypophosphatemia in infants, children, and adolescents may be due to hereditary or acquired disorders (see Table 543-1). Phosphate deficiency develops in patients receiving parenteral nutrition with inadequate amounts of phosphorus, in infants ingesting large quantities of phosphate-binding aluminum-containing antacids, and in very premature infants receiving only human breast milk.
Recently, the genetic basis of several disorders causing rickets has been elucidated. X-linked dominant hypophosphatemic rickets (XHR) results from a loss-of-function mutations in PHEX. This relatively common disorder (1:20,000 births) that causes rickets is discussed in detail in Chapter 542. Children with XHR should be comanaged with an experienced pediatric orthopedist who may prescribe braces. Progressive lower extremity deformities may benefit from hemiepiphysiodeses.24 Stature is short in the majority of children and adults with XHR. Beginning treatment with calcitriol and phosphate before 6 months of age may result in greater adult stature than when treatment begins after 1 year of age.25 The molecular basis of other genetic hypophosphatemic disorders are listed in Table 543-1 and eTable 542.1  .
.
Acquired and heritable disorders with renal tubular acidosis and Fanconi syndrome can also lead to phosphaturia and resultant rickets. These include cystinosis, tyrosinemia, galactosemia, Wilson disease, nephrotic syndrome, renal vein thrombosis, and mercury, lead, and copper poisoning.31
 HYPOPHOSPHATASIA
HYPOPHOSPHATASIA
Hypophosphatasia is a disorder of bone mineralization due to inactivating mutations of ALPL, the gene encoding tissue-nonspecific alkaline phosphatase.32 When function of this phosphomonoesterase is impaired, its endogenous substrates—pyrophosphate, pyridoxal 5′-phosphate, and phosphoethanolamine—accumulate, coat the surface of hydroxyapatite crystals, and restrict crystal growth.  The younger the clinically affected patient, the more severe are the manifestations of hypophosphatasia likely to be: (1) The perinatal form is usually lethal and includes marked osteopenia, which results in fractures of the extremities and ribs, chest wall deformities leading to respiratory insufficiency, cranial malformations and intracranial hemorrhage, and pyridoxine-dependent seizures. (2) The infantile form is recognized before 6 months of age and suspected because of impaired growth, hypotonia, deformities of the long bones and ribcage, large fontanelles and sutures with increased intracranial pressure, proptosis, papilledema, pyridoxine-dependent seizures, and radiographic evidence of rickets; almost half of these infants die of respiratory failure before age 1 year. (3) The childhood form has variable manifestations that range from premature shedding of deciduous teeth to rickets. (4) The adult form is often subclinical and found only when evaluating the parents of a child with hypophosphatasia or patients with a history of premature loss of teeth. (5) Odontohypophosphatasia is manifested only by premature shedding of primary teeth without clinical or radiographic evidence of rickets. (6) Pseudohypophosphatasia is phenotypically and biochemically similar to classic hypophosphatasia, but serum alkaline phosphatase activity in vitro is normal, indicating that enzyme activity upon artificial substrates is preserved, but that upon endogenous substrates is depressed.
The younger the clinically affected patient, the more severe are the manifestations of hypophosphatasia likely to be: (1) The perinatal form is usually lethal and includes marked osteopenia, which results in fractures of the extremities and ribs, chest wall deformities leading to respiratory insufficiency, cranial malformations and intracranial hemorrhage, and pyridoxine-dependent seizures. (2) The infantile form is recognized before 6 months of age and suspected because of impaired growth, hypotonia, deformities of the long bones and ribcage, large fontanelles and sutures with increased intracranial pressure, proptosis, papilledema, pyridoxine-dependent seizures, and radiographic evidence of rickets; almost half of these infants die of respiratory failure before age 1 year. (3) The childhood form has variable manifestations that range from premature shedding of deciduous teeth to rickets. (4) The adult form is often subclinical and found only when evaluating the parents of a child with hypophosphatasia or patients with a history of premature loss of teeth. (5) Odontohypophosphatasia is manifested only by premature shedding of primary teeth without clinical or radiographic evidence of rickets. (6) Pseudohypophosphatasia is phenotypically and biochemically similar to classic hypophosphatasia, but serum alkaline phosphatase activity in vitro is normal, indicating that enzyme activity upon artificial substrates is preserved, but that upon endogenous substrates is depressed.
The perinatal and infantile forms of hypophosphatasia are autosomal recessive disorders due to inactivating mutations in ALPL, whereas the childhood, adult, and odontohypophosphatasia forms may be transmitted as either autosomal recessive or dominant traits. Seizures occur in patients with loss-of-function mutations in ALPL because of deficiency of pyridoxine formed by dephosphorylation of pyridoxal 5′-phosphate; pyridoxine is a cofactor in the synthesis of the neurotransmitter γ-aminobutyric acid. The diagnosis of hypophosphatasia is established by the clinical and radiographic findings of rickets associated with low bone alkaline phosphatase activity and elevated serum levels of pyrophosphate and pyridoxal-5′-phosphate, increased urine excretion of pyrophosphate and phosphoethanolamine, and identification of the mutation in ALPL. No effective therapy for hypophosphatasia is currently available. Even small doses of vitamin D may cause hypercalcemia and should be avoided. Seizures may be responsive to pyridoxine.
 OTHER DISORDERS WITH LOW BONE MASS AND DECREASED MINERALIZATION
OTHER DISORDERS WITH LOW BONE MASS AND DECREASED MINERALIZATION
Juvenile Paget disease is characterized by muscular weakness, widened and bowed extremities, nontraumatic fractures of the long bones, kyphosis, and macrocephaly. Juvenile Paget disease is an autosomal recessive osteopathy characterized by rapidly remodeling woven bone, osteopenia, fractures, and progressive skeletal deformity. Serum alkaline phosphatase activity is substantially increased as are the levels of markers of osteoclast activity. This disease is the result of biallelic loss-of-function mutations of TNFRSF11B encoding osteoprotegerin, the decoy acceptor for RANKL, thereby enhancing osteoclastogenesis. Administration of recombinant osteoprotegerin to a child with juvenile Paget disease led to clinical and radiologic improvement.33,34 Familial expansile osteolysis clinically resembles juvenile Paget disease. There is increased bone turnover with medullary expansion of bone and thinning of cortices resulting in pathologic fractures and skeletal deformities; deafness and premature loss of dentition may also occur. This an autosomal dominant disorder due to a monoallelic activating mutation in TNFRSF11A encoding RANK, thus leading to augmented osteoclastogenesis.35
The metabolic bone disease that accompanies chronic renal failure (renal osteodystrophy) is discussed in Chapter 477.
 TREATMENT
TREATMENT
Specific treatment approaches are described above. Hypophosphatemia is treated with phosphorus supplementation, and if indicated, with calcitriol (1,25-dihydroxycholecalciferol). eTable 542.2  lists preparations of vitamin D and oral phosphate. Infants and young children prefer the soluble form of sodium-potassium phosphate; older children select a chewable phosphate tablet. Acidic potassium phosphate products are preferred, as they do not increase phosphate excretion while acidifying the urine, thus increasing the solubility of calcium phosphate. For infants and younger children, calcitriol is available as an oral solution at a concentration of 1 μg/mL. Routine monitoring of serum calcium, phosphate, creatinine, and parathyroid hormone (PTH) concentrations and urine calcium and creatinine excretion is essential. If hypercalcemia or hyper-calciuria (urine calcium excretion more than 4 mg/kg/day) occurs, the dose of calcitriol should be lowered. If serum PTH concentration increases above normal levels, the phosphate dose must be lowered. Maintenance of serum phosphate concentrations in the low normal range and alkaline phosphatase values within the high normal range are goals of therapy. Renal sonograms should be obtained prior to treatment and annually during therapy in order to detect early stages of nephrocalcinosis. Yearly skeletal radiographs to assess the degree of healing of the rickets are recommended, although complete radiologic healing of rickets is often difficult to attain (Table 543-2).
lists preparations of vitamin D and oral phosphate. Infants and young children prefer the soluble form of sodium-potassium phosphate; older children select a chewable phosphate tablet. Acidic potassium phosphate products are preferred, as they do not increase phosphate excretion while acidifying the urine, thus increasing the solubility of calcium phosphate. For infants and younger children, calcitriol is available as an oral solution at a concentration of 1 μg/mL. Routine monitoring of serum calcium, phosphate, creatinine, and parathyroid hormone (PTH) concentrations and urine calcium and creatinine excretion is essential. If hypercalcemia or hyper-calciuria (urine calcium excretion more than 4 mg/kg/day) occurs, the dose of calcitriol should be lowered. If serum PTH concentration increases above normal levels, the phosphate dose must be lowered. Maintenance of serum phosphate concentrations in the low normal range and alkaline phosphatase values within the high normal range are goals of therapy. Renal sonograms should be obtained prior to treatment and annually during therapy in order to detect early stages of nephrocalcinosis. Yearly skeletal radiographs to assess the degree of healing of the rickets are recommended, although complete radiologic healing of rickets is often difficult to attain (Table 543-2).
DISORDERS WITH LOW BONE MASS DUE TO IMPAIRED MATRIX FORMATION AND MINERAL DEPOSITION
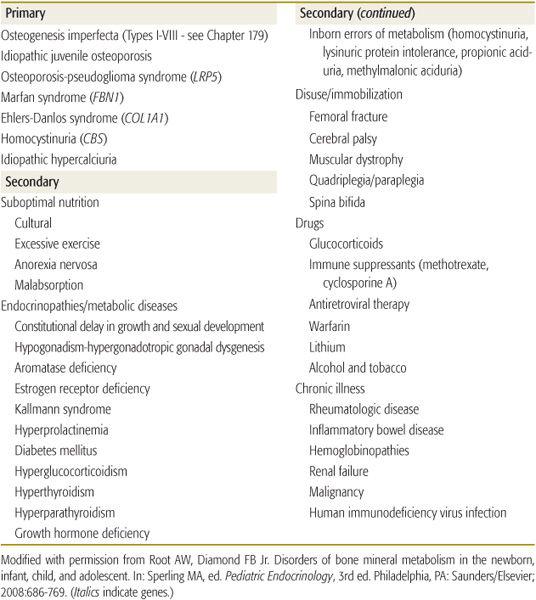
Although approximately 80% of bone mass is determined by genetic factors, exercise, nutrition, body weight and composition, and hormonal milieu modify attained bone mass and strength. Low bone mass may result from errors in the synthesis of collagen upon which calcium and phosphate are deposited or from lack of optimal amounts of mineral for deposition. Disorders associated with low bone mass due to impaired matrix formation and mineral deposition are listed in Table 543-3.36-58
 PRIMARY DISORDERS
PRIMARY DISORDERS
Osteogenesis imperfecta and other bone dysplasias are discussed in Chapter 179. Idiopathic juvenile osteoporosis is a disorder of low bone mass of unknown pathogenesis. It develops in mid- to late childhood and is associated with joint, muscle, and back pain; difficulty walking; shortening of the trunk; and kyphosis and compression fractures of the vertebrae and metaphyseal fractures in the long bones.59 Osteoblast activity and bone turnover are low, but peripheral chemical data are normal. Treatment is symptomatic. The disease often resolves during pubertal maturation. A monoallelic loss-of-function mutation in the gene (LRP5) encoding LDL receptor-related protein 5 has been detected in 15% of subjects.60
 SECONDARY DISORDERS
SECONDARY DISORDERS
Excessive consumption of carbonated cola beverages with a high phosphoric acid content that sequesters dietary calcium and diluted fruit juices has limited the intake of calcium to 55% to 70% of the recommended daily calcium allowances in school children.37,38 Non–weight-bearing sedentary activities (eg, video and computer games and television) and adiposity further impair bone mineralization.39 With severe malnutrition, as seen in anorexia nervosa, bone turnover rates and accrual are depressed, and total body, vertebral, and femoral neck bone mineral densities are significantly decreased.41-43
In chronically immobilized children (eg, cerebral palsy, spastic quadriplegia, muscular dystrophy), bone mass is low and femoral fracture rate is high during routine activities such as turning, dressing, or feeding due not only to lack of weight bearing but also to decreased nutrition, vitamin D and calcium intake, and to medications (eg, anticonvulsants, glucocorticoids).47,48 bone mineral density may be increased a bit in children with disabling cerebral palsy by administration of bisphosphonates or recombinant human growth hormone and even by assisted standing.49-51Table 543-3 also lists other chronic disorders often associated with decreased bone mass.63-70
 EVALUATION AND TREATMENT
EVALUATION AND TREATMENT
Active children with a history of frequent fractures with trauma most often do not have low bone mass unless there is a disease-associated risk factor (eg, cystic fibrosis, diabetes mellitus, celiac disease, inflammatory bowel disease, malignancy, long-term glucocorticoid exposure). Children are too often referred for evaluation of low bone mass because of erroneous analysis of DEXA data (use of t score rather than z score) or failure to consider the height, pubertal status, or bone age when interpreting bone mass data. Management of the child or adolescent with clear low bone mass is best achieved by effective treatment of the underlying disease if one is present and by ensuring adequate intake of calcium and vitamin D and consistent weight-bearing exercise in other subjects. The use of pharmacologic agents other than sex hormones in patients with hypogonadism and recombinant human growth hormone in patients with growth hormone deficiency is generally neither necessary nor recommended. Drugs that increase bone mass act either by inhibiting bone resorption (sex hormones, selective estrogen receptor modulators, calcitonin, bisphosphonates) or by stimulating bone formation (growth hormone, parathyroid hormone).
Table 543-3. Disorders with Low Bone Mass Due to Impaired Matrix Formation and Mineral Deposition
Bisphosphonates are analogs of pyrophosphate in which carbon is substituted for the oxygen that links 2 phosphate groups, to each of which is attached a carbon-side chain that enables the compounds to bind to and coat bone surface, thereby blocking its dissolution, and to impede osteoclast function directly.71,72 Bisphosphonates remain in bone for prolonged periods, and their effects are long lasting. In addition to osteogenesis imperfecta (OI), they have been used to increase mineralization and decrease fracture rate in children with glucocorticoid-induced osteoporosis, osteoporosis-pseudoglioma syndrome, Menkes disease, and cerebral palsy. Side effects of bisphosphonates are both acute (fever, myalgia, abdominal pain, vomiting, hypocalcemia) and chronic (inflammatory disorders of the eye, osteonecrosis of the jaw, and acquired aosteopetrosis.73 Paradoxically, overtreatment with bisphosphonates not only increases bone density but also fracture risk due to decreased bone strength.
Selection of a child for treatment with bisphosphonates requires careful consideration of the primary disease and whether in an individual patient the benefit-to-risk ratio is significantly positive. Presently, it is recommended that bisphosphonates be administered by physicians experienced in the care and management of children with low bone mass. Although parathyroid hormone (PTH) is primarily known to increase osteoclastogenesis and bone resorption, intermittent administration of small amounts of bisphosphonate or PTH1-34 (teriparatide) exerts a direct stimulatory effect upon the osteoblast, thereby increasing the rate of bone formation relative to that of bone resorption.74 Its effects in children with low bone mass have not been reported as yet.
DISORDERS WITH HIGH BONE MASS
Increased bone mass may be due to decreased resorption or increased formation of bone. Radiographically, high bone mass may be generalized or localized: osteosclerosis denotes thickening of trabecular bone; increase in the width of cortical bone is termed hyperostosis.75 Osteopetrosis is a disorder of high bone mineral density that is usually due to a defect in osteoclast formation or in its ability to reabsorb bone.76 The causes of increased bone mass are listed in Table 543-4.
 INFANTILE OSTEOPETROSIS
INFANTILE OSTEOPETROSIS
The autosomal recessive infantile or “malignant” form of osteopetrosis is manifested initially by failure to thrive and developmental delay and later by obstruction of cranial foramina leading to loss of sight and hearing and other cranial nerve functions as well as nasal obstruction due to choanal stenosis. Intense bone overgrowth results in pancytopenia, hepatosplenomegaly, sites of extramedullary hematopoiesis, increased susceptibility to infection and enhanced fracture risk because abnormal bone microarchitecture paradoxically results in decreased bone strength, mandibular and maxillary osteomyelitis, and death, often within the first several years of life.
Physical examination discloses a small infant with macrocephaly, nystagmus, and hepatosplenomegaly. Radiographically, there is relatively uniform increase in density of the skull, vertebrae, and axial skeleton; Erlenmeyer flask deformities at the distal ends of the long bones in older children; and alternating bands of sclerotic and lucent bone in the iliac wings. High bone mass is confirmed by DEXA.
Infantile osteopetrosis may be due to homozygous or compound heterozygous inactivating mutations in one of 3 genes: TCIRG1 encodes a subunit of the vacuolar proton pump necessary for transport of hydrogen ions (H+) from the cytosol into the resorption lacuna beneath the osteoclast; CLCN7 encodes a chloride channel required for movement of this anion into and acidification of the resorption lacuna; OSTM1 encodes a protein necessary for the normal posttranslational processing and stability of CLCN7.77-79 Management of the infant with osteopetrosis is symptomatic. In some infants with classical forms of osteopetrosis, bone marrow transplantation has halted progression of the disorder, albeit often with substantial residual deficits. In other patients, high doses of calcitriol and interferon-γ functioning as osteoclast-activating factors have been of some clinical benefit.
Table 543-4. Disorders with High Bone Mass



