Disorders of Bile Acid Synthesis
Peter T. Clayton
 METABOLIC PATHWAYS
METABOLIC PATHWAYS
The major bile acids produced in the liver are the taurine and glycine conjugates (amidates) of chenodeoxycholic acid (CDCA) and cholic acid (CA). Secretion of these conjugated bile acids into the canaliculi by the bile salt export pump fuels bile flow. They are powerful detergents. This property is important in keeping cholesterol in solution in bile, and for the digestion and absorption of fats and fat-soluble vitamins. The conversion of cholesterol to bile acids and the secretion of cholesterol into bile represent the major routes for elimination of excess cholesterol from the body. Thus, individuals with disorders of bile acid synthesis can present with liver disease due to impaired bile secretion (cholestasis), with gallstones, steatorrhea, symptoms of fat-soluble vitamin malabsorption, and buildup of cholesterol in tissues (causing neurological impairment, atherosclerosis, and xanthomata). 
The conversion of cholesterol to bile acids requires modifications to the sterol nucleus and to the sterol side chain.1 The major pathway for bile acid synthesis in adults (the “neutral” pathway) starts with conversion of cholesterol to 7α-hydroxycholesterol. A second pathway (the “acidic” pathway) starts with the conversion of cholesterol to 27-hydroxycholesterol, and the early steps can take place outside the liver. The neutral and acidic pathways share several enzymes; thus, inborn errors can disrupt both routes for the synthesis of bile acids. A simplified version of the major bile acid synthesis pathways is shown in Figure 165-1. 
CHOLESTEROL 7α-HYDROXYLASE DEFICIENCY
This disorder has not yet been detected in children. It is described in the online version of this chapter. 
3β-HYDROXY-Δ5-C27-STEROID DEHYDROGENASE DEFICIENCY
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Deficiency of 3β-HSD typically presents in the neonatal period with conjugated hyperbilirubinemia, elevated transaminases, and a normal γ-gutamyl transpeptidase.6,7 There is often associated steatorrhea. The liver biopsy shows a giant-cell hepatitis with evidence of cholestasis (retained bile pigment). Fat-soluble vitamin malabsorption may become evident from rickets (or even severe symptomatic hypocalcemia), vitamin K–responsive coagulopathy, or low plasma concentrations of vitamin E or vitamin A.6,8,9 If left untreated, the liver disease progresses with hepatomegaly, splenomegaly, and evidence of increasing fibrosis on a liver biopsy; the picture can resemble progressive familial intrahepatic cholestasis (PFIC), although pruritus is perhaps less common in 3β-HSD deficiency than in PFIC.10
 METABOLIC DERANGEMENT AND PATHOPHYSIOLOGY
METABOLIC DERANGEMENT AND PATHOPHYSIOLOGY
CDCA and CA synthesis are markedly impaired in 3β-HSD deficiency. Instead these patients synthesize considerable amounts of bile acids that resemble CDCA and CA, except that they retain the 3β-hydroxy-Δ5 configuration of cholesterol; the major urinary bile acids are sulfated 3β,7α-dihydroxy-5-cholenoic acid, sulfated 3β,7α,12α–trihydroxycholenoic acid, and their glycine conjugates. Transport of bile acids into bile is very much reduced and with it bile flow (cholestasis). Bile acid deficiency in the gut contributes to malabsorption of fat and fat-soluble vitamins. 
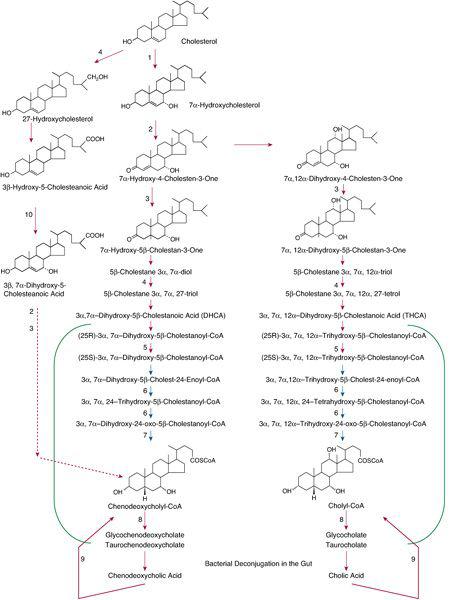
FIGURE 165-1. Simplified representation of two major pathways for the synthesis of bile acids from cholesterol—the acidic pathway on the left, starting with formation of 27-hydroxycholesterol, and the neutral pathway(s) on the right, starting with formation of 7α-hydroxycholesterol. Several enzymes participate in both pathways. Deficiencies have been described in cholesterol 7α-hydroxylase (1); 3β-hydroxy-Δ5-C27-steroid dehydrogenase/isomerase (2); Δ4-3-oxosteroid-5β-reductase (3); sterol 27-hydroxylase (4); α-methyl-acyl-CoA racemase (5); peroxisomal D-bifunctional protein (6); peroxisomal sterol carrier protein X (thiolase) (7); bile acid CoA: amino acid N-acyltransferase deficiency (8); bile acyl-CoA synthetase (9); and oxysterol 7α-hydroxylase (10). Reactions thought to occur predominantly in the peroxisomes are included in the green brackets. Disorders of peroxisome biogenesis affect many steps, including the reactions of peroxisomal β-oxidation, which is depicted by blue arrows.
 GENETICS
GENETICS
Deficiency of 3β-HSD is an autosomal recessive trait caused by mutations in the HSD3B7 (C27-3BETA-HSD) gene on chromosome 16p11.2-12.11
 DIAGNOSTIC TESTS
DIAGNOSTIC TESTS
Urine bile acid analysis shows peaks attributable to the sulfates and glycine-conjugated sulfates of 3β,7α-dihydroxy-5-cholenoic acid and 3β,7α,12α-trihydroxy-5-cholenoic acid. Plasma bile acid analysis shows the presence of these two bile acids and C27 analogues, particularly 3β,7α-dihydroxy-5-cholestenoic acid. The plasma concentrations of CDCA and CA are strikingly low for a child with cholestasis. 
 TREATMENT AND LONG-TERM OUTCOME
TREATMENT AND LONG-TERM OUTCOME
Emergency Treatments
Severe coagulopathy requires intravenous vitamin K. Seizures and tetany due to hypocalcemia require intravenous calcium and correction of vitamin D deficiency. Both of these problems will not recur once bile acid replacement has been started.
Bile Acid Replacement
The first patients were treated with chenodeoxycholic acid.13,8 This led to improvement in symptoms, rapid normalization of liver function tests, and improvement in the appearances in the liver biopsy. Follow-up has shown that treatment with CDCA fully controls the disease into adult life Some hepatologists advocate using cholic acid rather than CDCA.7
Δ4-3-OXOSTEROID 5β-REDUCTASE DEFICIENCY
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Δ4-3-oxosteroid 5β-reductase deficiency leads to increased urinary excretion of bile acids with a 3-oxo-Δ4 structure. This chapter will focus on the five patients with proven SRD5B1 mutations. 
All five patients presented as neonates with cholestatic jaundice with raised transaminases but normal γ-GT, low vitamin E, and prolonged clotting times that improved with parenteral vitamin K.15,16 Their liver biopsies showed giant cell transformation, canalicular and hepatocellular cholestasis, portal inflammation, septal fibrosis, occasional necrotic foci, and in some cases, increased extramedullary hemopoiesis. Cholestasis persisted in all cases. One infant progressed to liver failure and had a liver transplant at 19 weeks.15 One child failed to respond to ursodeoxycholic acid treatment but responded extremely well to treatment with chenodeoxycholic acid and cholic acid.15,22,23 One patient showed an initial response to chenodeoxycholic acid plus cholic acid but then required transplantation.15 Twin patients responded to treatment with cholic acid started at 8 months, and they were well at age 5 years.16
 METABOLIC DERANGEMENT AND PATHOPHYSIOLOGY
METABOLIC DERANGEMENT AND PATHOPHYSIOLOGY
The impaired function of the bile acid synthetic pathways results in very markedly reduced synthesis of CDCA and CA; these bile acids are present at unusually low concentration in the plasma and urine (for a child with cholestasis). The bile acid precursors with a 3-oxo-Δ4 structure undergo side chain oxidation to produce hydroxy compounds. The glycine and taurine conjugates of these bile acids are prominent in plasma and are the major bile acids in urine. Bile acids with a 3-oxo-Δ4 structure are not substrates for the bile salt export pump; hence the reduced bile flow (cholestasis). 
 GENETICS
GENETICS
Some cases showing a urine bile acid profile suggestive of 5β-reductase deficiency are due to homozygous mutations in the SRD5B1 (AKR1D1) gene (OMIM 604741). 
 DIAGNOSTIC TESTS
DIAGNOSTIC TESTS
Urine bile acid analysis shows that the main bile acids in the urine are 3-oxo-Δ4 bile acids and that excretion of the corresponding saturated bile acids is markedly reduced. Plasma bile acid analyses in infancy show markedly elevated concentrations of 7α-hydroxy-4-cholenoic acid and 7α, 12α-dihydroxy-4-cholenoic acid. In an older child, the major plasma bile acids are allo bile acids.23
 TREATMENT AND LONG-TERM OUTCOME
TREATMENT AND LONG-TERM OUTCOME
Treatment of a bleeding diathesis with parenteral vitamin K may be required. Vitamin D may be needed for rickets. Deficiency of 5β-reductase can progress rapidly to liver failure. However, treatment with bile acid replacement therapy can normalize liver function and lead to good long-term health. Successful regimes include chenodeoxycholic acid plus cholic acid and cholic acid alone. 
CEREBROTENDINOUS XANTHOMATOSIS (STEROL 27-HYDROXYLASE DEFICIENCY)
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
CTX has a range of clinical presentations, including cholestatic jaundice in early infancy,24-26 diarrhea and cataracts in the preschool child,27 learning difficulties in childhood, dementia in the teens/early adult life, and the development of tendon xanthomata and early atherosclerosis. The early liver disease can be fatal, and the early dementia can progress to crippling neurological dysfunction. These devastating outcomes are preventable with bile acid replacement therapy. For these reasons, early diagnosis, based on a high index of suspicion and appropriate tests, is extremely important. 
 METABOLIC DERANGEMENT AND PATHOPHYSIOLOGY
METABOLIC DERANGEMENT AND PATHOPHYSIOLOGY
Sterol 27-hydroxylase is present in the liver and in extrahepatic tissues. It provides a route for removing cholesterol from extrahepatic tissues.31 In CTX, accumulating cholesterol is converted partly to cholestanol. Sterol 27-hydroxylase also participates in the neutral pathway of bile acid synthesis.32 Hydroxylation of 5β-Cholestane-3α,7α,12α-triol does not occur in the C27 position; thus it accumulates in the liver. Consequently, it is metabolized by an alternative pathway, (in the endoplasmic reticulum) resulting in the synthesis of the characteristic bile alcohols that are found (as glucuronides) in the urine. Disruption of bile acid synthesis is probably responsible for the cholestatic liver disease of infancy, but the major symptoms of CTX in older children and adults are produced by accumulation of cholesterol and cholestanol in almost every tissue of the body. 
 GENETICS
GENETICS
CTX (OMIM 213700) is an autosomal recessive disorder caused by mutations in the CYP27A1 gene located on chromosome 2q33-qter. 
 DIAGNOSTIC TESTS
DIAGNOSTIC TESTS
Analysis of cholanoids (bile acids and bile alcohols) in urine shows a suggestive alcohol glucuronide pattern. Analysis of plasma shows that at all ages, the concentration of 5β-cholestane-3α,7α,12α,25-tetrol is increased. In adults, the plasma concentration of cholestanol is usually increased, but this is not a reliable diagnostic marker. 
 TREATMENT AND LONG-TERM OUTCOME
TREATMENT AND LONG-TERM OUTCOME
Without treatment, the neonatal-onset liver disease can be fatal. Adults with untreated CTX usually die from progressive neurological dysfunction or myocardial infarction between the ages of 30 and 60 years. The results of this treatment were first reported in 1984.34 A significant number of patients showed reversal of their neurological disability, with clearing of the dementia, improved orientation, a rise in intelligence quotient, and enhanced strength and independence. Urinary excretion of bile-alcohol glucuronides is markedly suppressed. Chenodeoxycholic acid almost certainly works by suppressing cholesterol 7α-hydroxylase activity; ursodeoxycholic acid, which does not inhibit the enzyme, is ineffective. Cholestatic liver disease in infancy may be self-limiting, but in those children in whom it is not, bile acid treatment has been successful; cholic acid is probably preferable to chenodeoxycholic acid.24,25
α-METHYL-ACYL-COA RACEMASE DEFICIENCY
This disorder is described in Chapter 162. Details of the impact on bile acid synthesis are given in the online version of this chapter. 
PEROXISOMAL D-BIFUNCTIONAL PROTEIN DEFICIENCY
This disorder is described in Chapter 162. Details of the impact on bile acid synthesis are given in the online version of this chapter. 
PEROXISOMAL STEROL CARRIER PROTEIN X DEFICIENCY
Details of this disorder, in particular its impact on bile acid synthesis, are given in the online version of this chapter. 
BILE ACID-COA: AMINO ACID N-ACYLTRANSFERASE
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
The author’s experience with patients who have proven mutations in the bile acid-CoA: amino acid N-acyltransferase (BAAT) gene is limited to observations on a 3-month-old infant who had cholestatic jaundice, vitamin D deficiency, and a liver biopsy showing mild portal and focal lobular hepatitis. Among the Amish, the presentation is with failure to thrive, sometimes with pruritus and occasionally with coagulopathy but not jaundice.50
 METABOLIC DERANGEMENT AND PATHOPHYSIOLOGY
METABOLIC DERANGEMENT AND PATHOPHYSIOLOGY
In BAAT deficiency, the liver produces unconjugated (nonamidated) bile acids instead of the usual glycine and taurine conjugates of CA and CDCA. The major urinary bile acid is unconjugated cholic acid. CA and CDCA are also excreted as sulfate and glucuronide conjugates.
 GENETICS
GENETICS
BAAT deficiency is an autosomal recessive disorder caused by mutations in the BAAT gene on chromosome 9q22.3. 
 DIAGNOSTIC TESTS
DIAGNOSTIC TESTS
The major urinary bile acid is unconjugated cholic acid. Other bile acids that may be detected include sulfated dihydroxycholanoic acid(s), trihydroxycholanoic acids, and their glucuronidated derivatives. 
 TREATMENT AND LONG-TERM OUTCOME
TREATMENT AND LONG-TERM OUTCOME
Treatment of vitamin K deficiency may be lifesaving, and treatment of rickets may require 1α-hydroxycholecalciferol or 1,25-dihydroxycholecalciferol. The Amish patients probably had improvement in symptoms with ursodeoxycholic acid  .50 The one patient monitored by the author had complete resolution of hepatitis in infancy with the help of ursodeoxycholic acid treatment and is asymptomatic at age 3 years.
.50 The one patient monitored by the author had complete resolution of hepatitis in infancy with the help of ursodeoxycholic acid treatment and is asymptomatic at age 3 years.
BILE ACYL-COA SYNTHASE DEFICIENCY
Bile acyl-CoA synthase (BACS) deficiency is a recently discovered disorder; details will be added to the online version of this chapter when the molecular basis is firmly established.
OXYSTEROL 7α-HYDROXYLASE DEFICIENCY
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
The first case of oxysterol 7α-hydroxylase deficiency was described in 1998 by Setchell and colleagues.51 This infant, who had severe cholestatic liver disease but normal γ-GT, did not respond to bile acid replacement therapy, required liver transplantation, and died of complications of the transplant.
Recently, mutations in the CYP7B1 gene encoding oxysterol 7α-hydroxylase have been shown in patients with hereditary spastic paraplegia type 5 (HSP5).52 The age of onset varies from 1 to 40 years. Thus, this seems to be another disorder that can cause cholestasis in the neonate and neurological disease later in life.
The age of onset varies from 1 to 40 years. Thus, this seems to be another disorder that can cause cholestasis in the neonate and neurological disease later in life.
 METABOLIC DERANGEMENT AND PATHOPHYSIOLOGY
METABOLIC DERANGEMENT AND PATHOPHYSIOLOGY
In the liver, oxysterol 7α-hydroxylase is essential for the acidic pathway of bile acid synthesis. Deficiency of the enzyme leads to synthesis of 3β-hydroxy-5-cholenoic acid  , which has cholestatic and hepatotoxic properties. In extrahepatic tissues, oxysterol 7α-hydroxylase contributes to a pathway for cholesterol degradation, and it provides the primary metabolic route for the modification of dehydroepiandrosterone neurosteroids in the brain. Loss of one or both of these functions may explain the neurological disease.
, which has cholestatic and hepatotoxic properties. In extrahepatic tissues, oxysterol 7α-hydroxylase contributes to a pathway for cholesterol degradation, and it provides the primary metabolic route for the modification of dehydroepiandrosterone neurosteroids in the brain. Loss of one or both of these functions may explain the neurological disease.
 GENETICS
GENETICS
Oxysterol 7α-hydroxylase deficiency is an autosomal recessive disorder caused by mutations in the CYP7B1 gene on chromosome 8q21.3. 
 DIAGNOSTIC TESTS
DIAGNOSTIC TESTS
In the reported neonatal case, 3β-hydroxy-5-cholenoic acid 3-sulfate and its glycine conjugate were excreted. In plasma, the main bile acids were 3β-hydroxy-5-cholenoic acid and 3β-hydroxy-5-cholestenoic acid, and the concentration of 27-hydroxycholesterol in plasma was very markedly increased. 
 TREATMENT AND LONG-TERM OUTCOME
TREATMENT AND LONG-TERM OUTCOME
The one reported patient who presented with cholestatic liver disease in infancy showed a deterioration with ursodeoxycholic acid and required a liver transplant for hepatic failure at 4.5 months. 
PEROXISOME BIOGENESIS DISORDERS
Disorders of peroxisome biogenesis are discussed in Chapter 162. As inborn errors affecting bile acid synthesis, they can be detected in infancy by analysis of urinary bile acids; this usually shows substantial excretion of taurine-conjugated tetrahydroxy cholestanoic acids. Analysis of plasma bile acids is a useful test for diagnosing an older child with a suspected peroxisomal disorder; the plasma typically contains the C27 bile acids, THCA and DHCA, and the C29-dicarboxylic acid. 
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


