Disorders of Accelerated Aging
John C. Carey
The syndromes associated with an accelerated or premature aging (often called progeroid) pheno-type represent an uncommon but medically and biologically important class of disorders. These conditions are characterized by physical manifestations that produce a prematurely aged appearance or symptoms suggesting accelerated aging unexpected for a child or adolescent. Although one of these conditions, Hutchinson-Gilford progeria (HGP), is the prototype for this community of diseases, there are at least 20 human syndromes that have been labeled as progeroid. It is a common mistake for both the clinician and the media to incorrectly lump all of these disorders under the heading of progeria, even though there are several discrete entities. Making an accurate diagnosis of the correct accelerated/premature aging syndrome is crucial because all of these conditions vary in inheritance pattern, natural history and prognosis. The plan of management and health care supervision is informed by the diagnosis here, as is the case of all genetic syndromes.
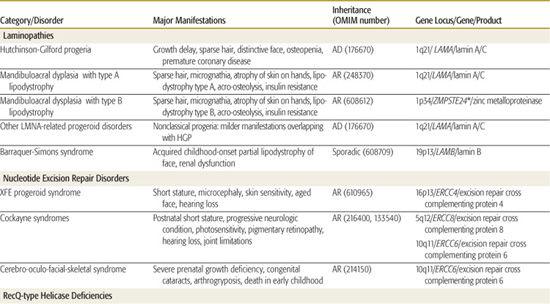
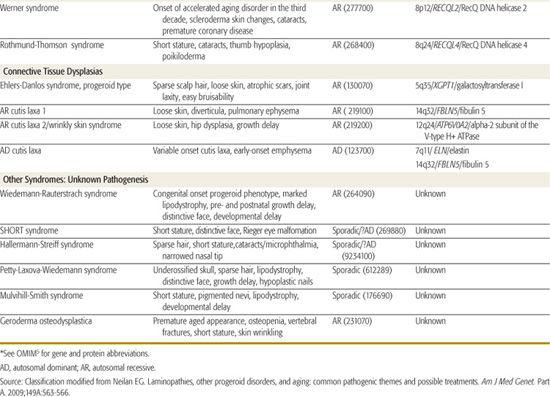
Table 184-1 (modified from a recent commentary by Neilan in 20091) classifies the commonly described progeroid syndromes by the pathogenetic category of the disorder. Hutchinson-Gilford progeria, the mandibuloacral dysplasias (MAD), and the connective tissue dysplasias with signs of premature aging will be discussed in more detail in subsequent section. Table 184-1 provides a listing of clinical manifestations of the conditions, as well as the inheritance pattern and the gene locus, gene name, and encoded protein if known. Of note, different mutations of the gene LAMA cause multiple different phenotypes, including HGP and a form of MAD as well as several nonclassical progeroid phenotypes1-3 displaying the genetic principle of allelic heterogeneity. It is also interesting that MAD is genetically heterogeneous (locus heterogeneity) and can be caused by mutations of LAMA or the gene ZMPSTE24, which encodes a peptide that is involved in the posttranslational modification of lamin A/C.4
DNA repair disorders (Table 184-1) account for some of the premature aging conditions, with the Cockayne syndromes representing the prototypes of this group. Given the postnatal onset of both HGP and Cockayne syndromes, investigators in the field are capitalizing on the knowledge of pathogenesis that arises from gene identification to develop therapies for these later-onset conditions, which is a logical progression given the advances in our understanding of the involved cell pathways. This line of reasoning would also apply to all of the conditions in Table 184-1, but those more serious conditions with later onset (eg, Cockayne, Werner, and HGP) represent particular priorities for this approach.
Table 184-1. Syndromes with an Accelerated Aging (Progeroid) Phenotype
GENERAL APPROACH
The reason that a particular syndrome is classified as an early or accelerated aging phenotype varies among all of the entities on the list. Often, the reason is related to facial and skin manifestations. What enters into the appearance of an aging phenotype is multifaceted: certain individual aspects of facial soft tissue and dermis change normally over time with age; there is increased wrinkling, loss of subcutaneous tissue, and alteration of the nasal and mandibular contour. The presence of some or all of these findings can contribute to a gestalt of accelerated aging. In males or females, hair loss and premature graying may contribute to a progeroid appearance. Any of these manifestations either singly or together can evoke an impression in the observer of premature aging. Thus, the inclusion of a condition in Table 184-1 is the result of the individual genetic condition’s intrinsic features affecting some or all of these manifestations. For example, the cutis laxas, the lipodystrophies, or the hair loss of HGP all account for individual reasons for the aged appearance. Perusal of the facial and cutaneous findings of each condition allows for distinction and understanding its inclusion in this grouping. The first step clinically is to ask why there is an impression of accelerated or premature aging. What specific components of the examination produce this impression: wrinkled skin, sparse hair, loss of subcutaneous tissue, or all of these manifestations?
In addition, some of the syndromes produce symptoms and signs unexpected for age; the premature coronary artery disease in HGP and Werner syndromes illustrates this concept.
Complete review of all 20 conditions is beyond the scope of this chapter. The Online Mendelian Inheritance in Man (OMIM) entry for each condition provides references and additional information on the clinical and genetic aspects.5
HUTCHINSON-GILFORD PROGERIA
The prototypic condition of early accelerated phenotype is the Hutchinson-Gilford progeria syndrome (HGP). This disorder was described independently by Hutchinson and Gilford toward the end of the 19th century.2 The syndrome is characterized by features of accelerated aging that begin during the first 2 years of life. Usually, affected infants appear normal at the time of birth, and the characteristic facies, hair loss progressing to total alopecia, and loss of subcutaneous tissue become apparent during the second and third years. The combination of lipodystrophy and hair loss comprise the characteristic appearance of accelerated aging in children with HGP. The term progeria, coined by Gilford, is derived from the Greek pro, meaning “early,” and geras meaning “old age.”2 Other features, also related to the appearance of an accelerated aging process, including stiffness of joints, osteopenia, and tightness of the skin over the trunk, develop in childhood and adolescence. Other facial characteristics, including the prominent forehead, absent eyebrows and eyelashes, smallness of the face compared to the skull, and absent ear lobule, add to the clinical appearance that allows for diagnosis. Notably, cognitive development is normal in individuals with HGP.
Death usually occurs due to complications of coronary arterial or cerebrovascular disease; the average lifespan is approximately 13 years. Hennekam comprehensively summarized the literature on over 130 cases in 2006.2 Many patients have been labeled as having HGP on a tenuous basis. Application of a stringent approach to phenotype, as outlined by Hennekam, allows for the recognition of this more discrete condition compared to the other overlapping or “nonclassical progeria” disorders. Now that the molecular and genetic basis of HGP is known to be owing to mutations in the gene LAMA encoding lamin A/C and diagnostic testing is generally available, diagnosis can be easily confirmed. Detection of the specific G608G point mutation in LMNA, a novel splice cite mutation, allows for diagnosis. Because the role of lamin A/C is now understood in cellular pathways, clinical trials designed to ameliorate the course of HGP are ongoing (http://www.clinicaltrials.gov).
The Progeria Research Foundation, the advocacy group for persons with HGP and related progeroid conditions, is a helpful resource for families (http://www.progeriaresearch.org).
RELATED LAMINOPATHIES
Mutations in the gene LMNA can be sequenced looking for mutations not only in HGP, but also in the related allelic disorders. More than 10 other conditions with mutations in the LMNA gene have been reported in the past decade.4 These conditions have been called laminopathies and include forms of Emery-Dreifuss muscular dystrophy, limb-girdle muscular dystrophy, the partial lipodystrophies (see Table 184-1), and a congenital and lethal skin disorder called restrictive dermopathy. Most important, some patients with mandibuloacral dysplasia syndrome (MAD), a condition with clinical manifestations overlapping those of HGP, have recessive mutations of LMNA. Patients with atypical Werner syndrome or progeria, conditions that overlap clinically with HGP and MAD have been reported to have mutations distinct from the typical G608G point mutation of HGP.2 Thus many accelerated early aging phenotypes are caused by changes of this important gene.
MANDIBULOACRAL DYSPLASIA
MAD was originally described in the early 1960s and a number of reported patients were originally labeled as HGP.2 More than 50 cases in several parts of the world had been redescribed as of 2000.6 Although there is overlap with HGP, the pattern is distinct and different. The presentation is usually later than it is HGP, with onset between ages 3 and 14. There is also a distinctively different face with a sharply defined and pointed nose and progressive micrognathia. As in HGP, there is a postnatal-onset short stature and progressive stiffness of joints. The acroosteolysis observed on radiographs is particularly helpful in the diagnosis of older children. The skin of the distal extremities and trunk becomes atrophic and subcutaneous fat is diminished in one of two patterns: In some patients, there is a generalized loss of subcutaneous fat (type B lipodystrophy), whereas others have a retention of fat in the neck and truncal areas with primarily loss of tissue in the limbs (type A).7 Most patients with MAD have a unique form of insulin resistance with a normal glucose tolerance test and hyperinsulinemia.
Some patients with MAD have mutations in the LMNA gene, whereas others have changes in the ZMPSTE24 gene, which is important in the post-translational modification of lamin A/C.4 Thus, the MAD phenotype may, in some situations be owing to the gene causing classical progeria (HGP), LMNA, whereas in others it is owing to changes in another gene in the same pathway. There are some clinical differences between the patients with the different mutations; most of the patients who have mutations of the ZMPSTE24 gene show the type B (generalized involving trunk and limbs) loss of subcutaneous tissue, whereas those with the more localized form have mutations of LMNA.
ACCELERATED AGING DISORDERS AND CONNECTIVE TISSUE DYSPLASIAS
One of the key manifestations that produces the impression of accelerated or early aging is loose or lax skin. Cutis laxa is a connective tissue dysplasia observed in a number of heritable conditions and syndromes and thus is etiologically heterogeneous; in all forms of cutis laxa, the skin of the face and/or trunk is loose or wrinkled. As summarized in Table 184-1, there are autosomal dominant and recessive forms of cutis laxa.5
An uncommon but relatively well-characterized condition is the progeroid type of Ehlers-Danlos syndrome (EDS). Although few cases are reported, this disorder falls into both the accelerated aging syndromes and the more classical connective tissue dysplasias in the Ehlers-Danlos grouping (Chapter 181). Patients with the EDS-progeroid type have loose skin and sparse scalp hair, giving the accelerated aging appearance, but also have atrophic scars, joint laxity, and easy bruis-ability as seen in classical EDS. The gene for this uncommon condition has been identified as the one encoding the galactosyltransferase I enzyme (Table 184-1).
OTHER SYNDROMES
The classical accelerated aging syndromes, HGP and MAD, do not present at birth. Three congenital disorders produce the accelerated or premature aging phenotype (Table 184-1). In all there is sparse hair, micrognathia, and changes of the face that resemble HGP. Hallermann-Streiff syndrome is the most well known; it can be distinguished by the congenital microphthalmia and cataracts. The Wiedemann-Rauterstrach and Petty-Laxova-Wiedemann syndromes, although both rare, should be considered when evaluating a newborn or infant with a progeroid phenotype.
REFERENCES
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


