Diseases of the Neuromuscular Junction
H. Royden Jones, Jr.
This chapter provides a practical approach, to the evaluation and treatment of the most common pediatric-acquired neuromuscular transmission disorder, myasthenia gravis. 
Myasthenia gravis (MG) is the most common neuromuscular transmission disorder in children and adults. Young children typically present with ocular MG manifested by ptosis and or diplopia.18Generalized MG presents with varied combinations of ocular, bulbar, neck, and limb weakness, and, rarely, respiratory distress, and is more common in adolescents.17Neonatal myasthenia gravis occurs in 15% of babies born to mothers with autoimmune MG.19 Congenital myasthenia gravis syndromes are a group of widely differing, much less-common neuromuscular transmission disorder, each characterized by compromised neuromuscular transmission safety margin.20 These genetically determined myasthenic disorders generally present during the first years of life with a more subtle ptosis and extraocular weakness. In contrast, other variants may have a very acute presentation, with the baby in extremis with a sudden life-threatening respiratory distress that very rarely leads to sudden death.20
 EPIDEMIOLOGY
EPIDEMIOLOGY
It is very unusual for autoimmune juvenile myasthenia gravis (JMG) to present before age one year.18 Although JMG is the most common pediatric neuromuscular transmission disorder, it occurs much less frequently than with adults. JMG incidence is 1.1 per 1 million children.21This accounts for 15% to 20% of MG patients in North America. There is a higher incidence in Chinese and Japanese children; JMG comprises 43% to 60% of mainland Chinese myasthenia gravis patients, as well as 60% of MG patients in Hong Kong.22,23 Here, the incidence of ocular MG is 10 times more frequent than generalized JMG. 
JMG children rarely have other autoimmune disorders.8 A few examples include pauciarticular juvenile chronic arthritis,23 Hashimoto thyroiditis,28 and systemic lupus erythematosis.33 A 1983 JMG review identified 14 of 149 children with associated autoimmune disorders that included rheumatoid arthritis (5), juvenile-onset diabetes mellitus (3), asthma (3), and thyroid disorders (3).34A late-onset multiple sclerosis is also reported.35
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
The clinical hallmark differentiating juvenile myasthenia gravis (JMG) from other neurologic disorders is its predominant fluctuating course with diurnal variation. Initially, JMG patients typically awaken feeling normal, only to experience late-day symptomatology, for example, ptosis, dysphagia, dysarthria, and or fatigue. Although a few other neuropathologic entities may have intermittent symptoms, none develop the archetypic daily diurnal JMG fluctuations. Rarely in adolescents, the differential diagnosis includes transient ischemic attacks, minor focal seizures, and multiple sclerosis in adolescents.
Parents are often first concerned when they note a drooping eyelid late in the day. A false sense of security often occurs, when symptoms significantly improve or totally clear the next morning. This leads parents or physicians to inappropriately dismiss their concern or suggest this was just fatigue from “overexercising.” Once ptosis becomes better defined, diplopia develops, and both soon become more persistent. Occasionally, JMG presents primarily with dysphagia and/or dysarthria, as well as a more generalized weakness. At its most severe degree, the inherent muscle weakness of JMG occasionally leads to early respiratory compromise.
The two most important clinical observations supporting a JMG diagnosis are a late day appearance and/or worsening (ie, diurnal) and a fluctuating clinical course (“here today gone tomorrow”). Although the symptoms initially clear transiently, relatively soon children with JMG develop a more established disorder.
When the JMG patient develops more generalized symptoms, these are typically a diurnal appearance of weakness of both masticators and facial muscles manifested by jaw and lip fatigue with a repeated tendency to stop chewing or drooling part way through a meal; if severe, the child is unable to complete the meal. Occasional children experience diurnal difficulty sipping with a straw, and speech becomes “breathy.” Sometime a brief rest permits the child to resume the meal, start speaking more easily with clearer articulation, and repair any tendency to nasal regurgitation or breathy speech. If the child has previously learned to whistle he/she will often note the loss of this function. As proper function of the velum (soft palate) pharyngeal valve is essential for unrestricted respiration, effective deglutition, and normal speech, weakness of the muscles contributing to valve closure may be the presenting complaint of JMG.
Rarely, juvenile myasthenia gravis (JMG) primarily presents in a fashion mimicking a classic myopathy with proximal more than distal extremity muscle weakness.21,28,34,37,38 There are no preceding or accompanying ocular or bulbar symptoms, although diaphragmatic and intercostal muscle weakness may occur, compromising breathing. If severe enough, a respiratory, myasthenic, crisis follows. Patients with neuro-muscular transmission disorders do not develop significant headaches or neck pain unless related to skeletal muscle fatigue; however, these clinical accompaniments must initially alert the physician to the possibility of a primary spinal or intracranial process. Similarly, a JMG diagnosis is untenable when patients demonstrate pupillary or sphincter dysfunction.
The accurate assessment of strength, particularly demonstration of fatigability, is often difficult in young children. Even when one is examining an older child, the physician may be challenged to physically demonstrate support for symptoms of subtle and fluctuating weakness. When these are not yet specifically substantiated, a late afternoon examination of the patient may define a mild degree of weakness not present earlier in the day. One must look for signs of subtle neuromuscular compromise, such as minor degrees of ptosis, trouble sipping with a straw, fatigue counting numbers, tendency for the neck to droop secondary to neck flexor weakness, inability to do push-ups (related to triceps muscle weakness), or inability to climb stairs. Details of the neurological exam are discussed in further on the DVD. 
 DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
Usually, diagnosis of juvenile myasthenia gravis is very straightforward; however, on other occasions a skillfully obtained neurologic history and exam are required to diagnose juvenile myasthenia gravis (JMG). The major two critical clues include (1) diurnal fluctuation and (2) disproportionate fatigability. Newly developed ptosis offers a major diagnostic key to appropriate diagnosis, especially when there is diurnal variation. Pupillary responses are normal in myasthenia gravis (MG). If the pupil is dilated, especially with associated ptosis, a third cranial nerve palsy, and not JMG is present. A miotic (ie, constricted) pupil with minimal ptosis and subtle ipsilateral decreased facial sweating defines the Horner syndrome, secondary to sympathetic fiber damage. Other diagnoses to be considered include the following. Parasellar lesions, for example, craniopharyngiomas or pituitary tumors, and medulloblastomas. The lack of diurnal variability or daily fluctuations makes JMG unlikely in these various settings. 
The acute-onset Miller Fisher variant of Guillain-Barre syndrome presents with ophthalmoplegia, ataxia, and areflexia. There is no diurnal variation or fatigability.
Congenital and mitochondrial myopathies and some muscular dystrophies frequently present with ptosis and limited extraocular muscle weakness function.20 However, there is no diurnal variation or diplopia, and the ptosis is more subtle and does not have a relatively acute onset or significant asymmetry.
Rarely, children with JMG present with proximal leg muscle weakness or a floppy neck with cervical muscle weakness.45,46 Differential diagnostic considerations here include spinal muscular atrophy, chronic inflammatory demyelinating polyneuropathies, Lambert-Eaton myasthenic syndrome, and dermatomyositis. Early on, one must vigorously avoid making the inappropriate diagnosis of an anxiety disorder or a conversion disorder.47
 DIAGNOSTIC EVALUATION
DIAGNOSTIC EVALUATION
Although there are multiple JMG diagnostic modalities,48,49testing for the presence of antibodies directed against the acetylcholine receptor is the most definitive study.11 These results are not immediately available in the acute care setting and are not always positive. More traditional means, such as edrophonium chloride (Tensilon) and EMG are very useful.35,50 An ice water pack test is gaining increased use for the child with ptosis.51-53 However, in contrast to Tensilon, the ice water test has very limited applicability for studying other muscles. EMG continues to be one of the two primary means of diagnosing JMG in the acute setting, particularly in the generalized form. When the child has very mild symptoms and thus time permits, especially when one is able to obtain a positive ice pack test leading to a preliminary diagnosis of JMG, there is an opportunity to wait for the definitive antibody study results.11 Because there is an association between myasthenia gravis and thymoma, all new-onset JMG patients need to have chest CT scanning.
Acetylcholine Receptor Antibodies
Antibody testing is the gold standard and most specific diagnostic modality for autoimmune juvenile myasthenia gravis (JMG); patients with positive results are referred to as antibody seropositive.11,54 These acetylcholine receptor antibodies (AChRAbs), derived from the patient’s serum, bind directly with other skeletal muscle acetylcholine receptor (AChR)’s. Their effects at the AChR are multifaceted. These effects include an accelerated AChR degradation, cross-linking, endocytosis, a functional blockade, and a complement-mediated lysis of end plates. In adults antibodies are detected in 10% to 50% of primary ocular and 90% of patients with generalized myasthenia gravis. However, seropositivity is less common in JMG. Fifty percent to 64% of prepubertal patients are seropositive, compared to 68% to 82% of peripubertal patients and 91% of postpubertal adolescents with acquired myasthenia gravis.55-58
The relatively high frequency of seronegativity in younger, presumed JMG children creates special problems. These are the very patients in whom discrimination between seronegative autoimmune JMG and congenital forms is most important. Pediatric patients are often too young to accurately report symptoms. The natural tendency to have frequent daytime naps and the pediatrician’s inability to perform a precise clinical examination in the younger child make a positive clinical diagnosis of autoimmune JMG most difficult. Repeated AChR antibody measurements, over a few years, occasionally demonstrate seronegative to seropositive conversion.35 Also, since AChRantibody levels may be lower in prepubertal patients than in older patients, experienced laboratories need to be used for accurate serologic diagnosis.11
When the testing for acetylcholine receptor (AChRAb) binding antibodies is negative, at least 2 additional AChRAb assays are available.13 These include “modulating” antibodies that bind selectively to exposed segments of AChR on living muscle, increasing their degradation, as well as “blocking” antibodies that block the acetylcholine binding at the receptor. The modulating antibody provides occasional help for the presumed juvenile myasthenia gravis child who is seronegative for the conventional AChRAb assay.54The blocking antibody, although available, has not been of clinical value.
Anti-Muscle-Specific Kinase Antibodies
This antibody is present in approximately 50% of anti-acetylcholine receptor antibody seronegative adult myasthenia gravis (MG)55-58 as well as occasionally in juvenile myasthenia gravis (JMG).59-61 Current literature suggests that muscle-specific kinase positivity is more likely with the most significantly affected JMG patients with severe generalized weakness.60,61
Low-Affinity Anti-Acetylcholine Receptor Cluster–Binding Antibody
This newly defined antibody occurs in 50% of MG patients who do not have positive tests for acetylcholine receptor antibodies (AChRAb’s) or anti–muscle-specific kinase antibodies.63 There is no data currently available for children,64 but this newly defined study may possibly become useful when seemingly classic JMG patients are seronegative.
Ocular Ice Pack Test
Ptosis is the most common JMG finding. It is often more apparent during hot weather; in contrast, myasthenia manifestations are improved by cold. Placement of an ice pack over an affected eye, the ocular ice pack test (OIPT), is a very logical initial test, even before attempting the use of edrophonium.51-53 The OIPT is entirely benign. An ice pack is gently applied over the ptotic eye. MG patients usually experience a rapid eyelid elevation, usually within 5 minutes.51 A major advantage of OIPT is that it negates need for an intravenous injection (see below). An OIPT is particularly helpful in relatively mildly affected patients and is becoming the initial standard test for JMG with ptosis. The OIPT has a high sensitivity; 8 of 10 MG patients showed improvement, whereas no controls improved.51
Acetylcholinesterase Inhibitors
Acetylcholinesterase inhibitors are pharmaceutical agents that specifically block the enzymatic breakdown of acetylcholine by acetylcholinesterase, leading to varying degrees of prolongating the effectiveness of acetylcholine at the postsynaptic neuromuscular junction.
Edrophonium Chloride (Tensilon) Edrophonium is used less frequently now due to (1) the effectiveness of ocular ice pack test, (2) its potential cholinergic side effects, and (3) the discontinuation of its manufacture. However, another pharmaceutical company (Bioniche Pharma) is now returning the substance to production as Enlon. The standard childhood testing is initiated by an intravenous test dose of 0.01 mg/kg of edrophonium. If this is well tolerated, the dose may then be increased to 0.1 mg/kg. The maximum accepted dose is 0.15 mg/kg or no more than a total of 10 mg for a mature adolescent.65
Edrophonium has a rapid onset of action, occurring within 1 to 2 minutes. Its effect is short lived, usually disappearing within 5 minutes. An unequivocal improvement in an objectively monitored muscle, such as the eyelid, extraocular muscle, or voice, is considered a positive response. A ptotic eyelid is the primary clinical finding best suited for edrophonium evaluation. Positive responses are unequivocal  . Side effects are primarily muscarinic, including increased salivation, lacrimation, gastrointestinal cramping and even diarrhea, miosis, and fasciculations. The older child may feel presyncopal. More major side effects are related to potential cardiac rhythm or respiratory compromise. These are primarily of concern in older adults. Judicious delivery of the medication instead of a direct full push is always recommended; this delivery usually prevents these side effects. Atropine (0.5 mg) must be immediately available for intravenous injection, as it will promptly reverse these cholinergic symptoms. Concomitant EKG monitoring is recommended.
. Side effects are primarily muscarinic, including increased salivation, lacrimation, gastrointestinal cramping and even diarrhea, miosis, and fasciculations. The older child may feel presyncopal. More major side effects are related to potential cardiac rhythm or respiratory compromise. These are primarily of concern in older adults. Judicious delivery of the medication instead of a direct full push is always recommended; this delivery usually prevents these side effects. Atropine (0.5 mg) must be immediately available for intravenous injection, as it will promptly reverse these cholinergic symptoms. Concomitant EKG monitoring is recommended.
Neostigmine The major value of neostig-mine is its availability for either subcutaneous or intramuscular administration. The recommended intramuscular dose is 0.15 mg/kg.17 When administered intravenously, the dose must be reduced to 0.05 mg/kg. This medication requires a longer time (15 minutes) for clinical response than does edrophonium. Its longer duration of action of 1 to 4 hours provides more time to observe the response specifics. Neostigmine requires similar cautions and has similar side effects to edrophonium.
Pyridostigmine (Mestinon) On occasion, when no diagnostic study is positive, a pyridostig-mine therapeutic trial may be diagnostically helpful. Pyridostigmine is available in 60-mg tablets best given 45 to 60 minutes before meals. One 60-mg tablet is a good starting level for adolescents, with an adjustment downward according to the child’s age and size. Special caution is indicated with patients having bronchial asthma.
Slurp Test
When children with juvenile myasthenia gravis present with swallowing and speech difficulty, the physician needs to easily quantitate this function diagnostically Usually, it is very difficult to quantitate pharyngeal dilator, pterygoid, hypoglossal, and tongue muscle function. The slurp test is a very practical means to assess function of the lower bulbar musculature, an area that characteristically is vulnerable to juvenile myasthenia gravis fatigue.65 The child is provided a straw and encouraged to rapidly drink the entire contents of a 4-oz cup and in so doing makes a loud slurping sound as the drink is finished. The duration of time from start to the production of the characteristic sound is measured. Healthy children demonstrate median baseline slurp test times ranging from 4 to 18 seconds, with younger children tending toward the longer times. Once a specific baseline is established for a child presumed to have juvenile myasthenia gravis, the slurp test is used to assess responsiveness to anticholinesterase inhibitors or as a guide to decline in bulbar function when concerned about an impending myasthenic crisis.
Electromyography
EMG was the standard means used to diagnose juvenile myasthenia gravis prior to the development of specific antibody studies.66-68 This neurophysiologic study continues to provide an important role, particularly with the acutely ill child in whom rapid diagnosis is important, because antibody studies are not available for at least a few days. In the acute setting, the Guil-lain-Barre syndrome is part of the differential diagnosis. Such patients typically have marked slowing of motor nerve conduction, totally different from juvenile myasthenia gravis.
Nerve conduction studies are typically normal in juvenile myasthenia gravis, with the exception that in severe generalized disease the motor response may be of low amplitude. Repetitive motor nerve stimulation in juvenile myasthenia gravis demonstrates abnormal muscle fatigue secondary to loss of the safety factor typically generated at the normal neuro-muscular junction.
 TREATMENT
TREATMENT
The therapeutic approach to the initial and ongoing juvenile myasthenia gravis care depends on whether the child has primary ocular disease (grade I) or generalized myasthenia gravis (grades II to IV). An algorithm for juvenile myasthenia gravis therapy was previously developed.14 This is modified in Table 571-1. The need to invoke use of thymectomy and or corticosteroids is dependent on the severity and the acuity of the juvenile myasthenia gravis or a poor response to initial symptomatic therapy. This algorithm is not a specific outline for every child, as each case requires precise individual direction. Therefore, it is emphasized that this table serves as an overview and introduction to this chapter’s therapeutic section.
Two different medication classes are appropriate for juvenile myasthenia gravis therapy. Cholinesterase inhibitors usually provide initial symptomatic control in the mildly affected child. A short-term course of these drugs may bring juvenile myasthenia gravis into total remission. This approach may also suffice for grade I, ocular juvenile myasthenia gravis children.
Achievement of therapeutic immunosuppression in a child with generalized juvenile myasthenia gravis usually requires immunomodulatory medication, with just a few exceptions, as noted in the previous paragraph. These usually provide clinical stabilization and, it is hoped, a cure. There is never any urgency to discontinue or decrease successful immunosuppressive therapy as juvenile myasthenia gravis almost always requires at least a few years to be brought into complete remission. As juvenile myasthenia gravis and adult myasthenia gravis are very rare chronic autoimmune disorders, it is best for the experienced neuromuscular neurologist to maintain primary therapeutic responsibility. This will often avoid premature medication withdrawal and its potential for recurrent exacerbations.
Symptomatic Therapy
Pyridostigmine (Mestinon) Pyridostigmine is the primary cholinesterase inhibitor used to provide early symptomatic relief. It is has just a 2- to 4-hour effect. The pediatric dose is 0.5 to 1.0 mg/kg every 4 to 6 hours. It is available in 60-mg tablet formulation or a syrup form that has a concentration of 60 mg/5 mL.14 A longer-acting form of Mestinon is useful at bedtime when symptoms develop during sleep or are problematic on awakening; with rare exceptions, this is not needed. The total maximum daily dose of pyridostigmine should not exceed 720 mg/day for a fully grown adolescent and be modified accordingly for the younger child.
Neostigmine When parenteral administration of anticholinesterase medication is necessary for symptomatic control, particularly when swallowing is impaired, thus making it impossible to take pills, neostigmine may be substituted for pyridostigmine with a dosage of 0.3 mg/kg.
Immunomodulatory Medications
Corticosteroids Prednisone is the steroid primarily used. It is wise to begin with a small dose because of the known paradoxical increased weakness that occurs when high doses are initially employed for myasthenia gravis.71 In a mature adolescent, a 5–10 mg dose is given during the first few days, and the total dosage is gradually increased by 10 mg every few days in a hospital setting or weekly in an outpatient environment, eventually achieving a dose of 1 to 2 mg/kg/day. A remission usually occurs within 2 to 6 months.
Prednisone is fraught with many concerns for a child, particularly growth retardation. Thus plasmapheresis or intravenous immunoglobulins are considered before prednisone if symptomatic control is not achieved with pyridostigmine. Methylprednisolone may also be used to establish an initial remission or for a sudden unexpected clinical deterioration in a patient previously in remission.14 In the adolescent, 250 to 750 mg given intravenously over 2 to 4 hours may be considered. The response can be quite varied, from a total remission to no improvement.14 Once stabilized, the issue arises as to whether to move to thymectomy or add corticosteroids sparing agents.
Azathioprine This medication is a cytotoxic agent metabolized to 6-mercaptopurine. Its major side effects are liver toxicity and myelosuppression in 10% of patients. The initial recommended dose is 1.0 mg/kg/day, with weekly 0.5-mg dose increments to a maximum of 2.5 mg/kg/day.14 The primary reason that azathioprine is not used as the initial immunosuppressive agent is the long delay in onset of action, with 6 to 12 months usually being required for clinical effectiveness. However, if corticosteroids have already been initiated effectively, azathioprine is started as soon as the corticosteroid-induced remission occurs. Eventually, this can become the primary immunomodulatory therapy as the prednisone is very slowly tapered on an alternate-day basis.
Table 571-1. Therapeutic Options for Juvenile Myasthenia Gravis
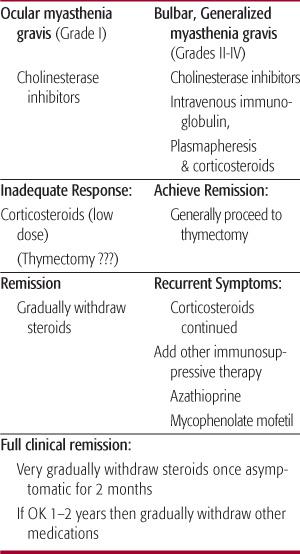
Mycophenolate Mofetil This immunosuppressive agent was initially heralded as having a faster onset than azathioprine, but it has no more effect when given with prednisone than when compared with use of prednisone alone.72 Side effects include gastrointestinal, hematologic, and dermatologic issues.
Cyclophosphamide Cyclophosphamide was the most efficacious medication in a 1990 study of juvenile myasthenia gravis from Warsaw. Ten of 20 children received cyclophosphamide, 17 prednisone, and 11 azathioprine.70 Although cyclophosphamide traditionally is considered to have too many toxic side effects,  this medication needs further consideration in the management of juvenile myasthenia gravis.
this medication needs further consideration in the management of juvenile myasthenia gravis.
Tacrolimus and Cyclosporine A Cyclosporine A has been used as a steroid-sparing agent for adult myasthenia gravis.76,77 It has not specifically been studied in children.14 In adult myasthenia gravis treatment trials show improved strength and a reduction of corticosteroid doses within the first 2 months of therapy.76,77 The typical dose is 5 mg/kg/day given in 2 oral doses 12 hours apart.14 Tacrolimus (FK-506) is another immunosuppressive agent recently trialed for treatment of juvenile myasthenia gravis.73,74 It has led to symptom remissions in 2 weeks in case reports without accompanying side effect.73 In a large adult series of tacrolimus-responsive myasthenia gravis, 87% of patients achieved a pharmacologic remission.72 Complications of both of these medications should be monitored as in patients following whole organ transplantation (Chapter 128).
Limited-Term Immunomodulatory Therapies
When treating grade III to IV juvenile myasthenia gravis patients, it is not necessary to push large doses of acetylcholinesterase inhibitors with their potential for cholinergic crisis. When confronted with a seriously ill child with juvenile myasthenia gravis, 2 forms of short-term treatment are available to effect a rapid clinical improvement. Plasmapheresis and intravenous immunoglobulin provide a means for either allowing a thymectomy to be performed after the child achieves a maximal improvement or to allow time for corticosteroids to be used and gain remission.
Plasmapheresis Plasmapheresis, or plasma exchange, removes antibodies and other proteins from the plasma. Plasmapheresis is rapidly effective; clinical improvement usually occurs within a few days to a week.78,79It is best used both at the initiation of immunosuppressive therapy providing rapid control until the various medications become effective, and prior to thymectomy, thus maximizing the child’s clinical state and diminishing perioperative morbidity. A course of 3 to 5 plasma exchanges provides clinical relief for 1 to 2 months. This treatment modality is equally effective in antibody-positive, muscle-specific kinase antibody–positive, and antibody-negative patients. In fact in anti–muscle-specific kinase antibody–positive individuals plasmapheresis may be the only effective therapy. The most common complications were fever (7.7%), urticaria (7.4%), and hypocalcemic symptoms (7.3%).
Intravenous Immunoglobulin In a severely ill child with juvenile myasthenia gravis, intravenous immunoglobulin is one of the most useful modalities to achieve a short-term remission. The usual dose is 2 g/kg given as 0.4 gm/kg aliquots daily over 5 days, but it can be given in 1 to 2 larger doses. More specific studies of intravenous immunoglobulin are needed in juvenile myasthenia gravis.
Thymectomy This has been one of the traditional therapies for myasthenia gravis ever since Blalock’s serendipitous observations of its possible positive effects on the course of myasthenia gravis.83 However, it has been extremely difficult to interpret the effect of thymectomy in juvenile myasthenia gravis, as is also the case in adult myasthenia gravis.83-88 Reports as to the effectiveness of thymectomy vary, with 50% to 90% showing improvement and 11% to 75% showing remission.14,86
Understandably, there is serious concern about the performance of thymectomy in the young child with a potentially immature thymus, thus leading to immunoincompetence. Infants often undergo complete or partial thymectomy during surgery for congenital heart and there is no difference in infection rates, although the sequelae remain controversial.86 There is no difference in the reported frequency of malignancy following thymectomy for childhood myasthenia.36,86 A particularly good response to early thymectomy among peripubertal patients treated within 1 to 2 years of disease onset occurred in 2 retrospective analyses.28,86,89,90
Contraindicated Therapies
The neuromuscular junction is susceptible to a number of medications that negatively affect neuromuscular transmission. Certain neuro-muscular blocking agents must be particularly avoided, including succinylcholine, d-tubocurarine, and vecuronium. Care must also be taken with cardiac medications such as quinine, quinidine, or procainamide, as well as calcium channel blockers and beta-blockers, including propranolol, and timolol maleate eyedrops.
Certain antibiotics, particularly kanamycin, neomycin, streptomycin, tobramycin, gentamicin, colistin, erythromycin, and ciprofloxacin may also interfere with neuromuscular transmission. Other antimicrobials also need to be avoided or monitored closely with juvenile myasthenia gravis patients, particularly including the fluoroquinolones, namely norfloxacin, ofloxacin, and pefloxacin. Magnesium salts such as Milk of Magnesia, Maalox, and Epsom salts, as well as iodinated contrast agents must be used judiciously if at all.
D-penicillamine may induce an autoimmune response at the neuromuscular junction in its own right, thus mimicking myasthenia gravis. This should never be used in myasthenic patients. A number of other medications may exacerbate the degree of weakness observed in some juvenile myasthenia gravis patients. Whenever any juvenile myasthenia gravis patient receives a new medication, it is imperative that the physician and the child’s family be alert to any potential deleterious effects manifested by increased weakness.
Generalized Juvenile Myasthenia Gravis
The best-detailed study of prognosis in generalized juvenile myasthenia gravis is a 40-year Mayo review in 1983.36 Only a small percentage of the children received immunosuppressive therapy. One hundred forty-nine juvenile myasthenia gravis children were studied from onset of the disease, with a minimum follow-up of 4 years and a median follow-up of 17 years. Of the more severely affected children, 85 (57%) had a thymectomy. A spontaneous remission rate of 22.4 per 1000 person-years was observed, regardless of disease duration. During the first year after a thymectomy, the remission rate was 260 per 1000 person-years.36
More recently, prognostic information has been gathered on juvenile myasthenia gravis patients who had more sustained immunosuppressive therapy. A study from India reviewed 77 juvenile myasthenia gravis children over a 34-year period (1965–1999).47 Average age of onset was 8 years, with a 6-year follow-up. Ocular (grade I) juvenile myasthenia gravis was found in 30% of children at onset; only 3 had primary limb girdle juvenile myasthenia gravis. Corticosteroids were provided to 67% and azathioprine was added to 7% of these children. At follow-up, 25 children were asymptomatic, 10 achieved a complete remission, 28 had a partial improvement, 9 were either unchanged or worse, and 2 died. The authors suggested that juvenile myasthenia gravis patients have a more benign course, better long-term outcome, and a much lower association with thymoma.47
Ocular Juvenile Myasthenia Gravis
In a study of patient with ocular juvenile myasthenia gravis in Turkey, only 1 of 15 ocular juvenile myasthenia gravis patients developed grade II generalized juvenile myasthenia gravis, with the other 14 maintaining their primary ocular presentation over a 2- to 24-year follow-up. An exacerbation occurred in just 3 patients 5 to 11 years after onset.94 In a Toronto study of juvenile myasthenia gravis, 14 of the autoimmune cases had ocular signs and symptoms; 5 of these progressed to generalized juvenile myasthenia gravis within 1 to 23 months. The 9 ocular myasthenia cases were treated primarily with pyridostigmine; 2 of these also received prednisone. One California study of primary ocular juvenile myasthenia gravis, beginning before age 12 years, demonstrated a median age of presentation with extraocular muscle weakness and ptosis at 26 months. Pyridostigmine therapy sufficed in all but 6 of 21 children. A complete resolution free from medication occurred in 4 of 21 children. Progression to generalized juvenile myasthenia gravis occurred in just 3 of 21 children during a 2- to 15-year follow-up.96 This is at a much lower rate than in adults.
TRANSIENT NEONATAL MYASTHENIA GRAVIS
Circulating maternal IgG acetylcholine receptor antibodies normally cross the placenta to enter the fetal circulation where they may bind at the fetal neuromuscular junction acetylcholine receptor. Although all infants born to seropositive mothers have elevated circulating acetylcholine receptor antibodies, only about 15% of these babies develop clinical signs of transient neonatal myasthenia gravis (TNMG). These autoantibodies may persist for 4 to 6 months postpartum.97 If an affected mother has one TNMG infant, her subsequent infants have a higher likelihood of being similarly affected.98,99
Typical symptoms develop shortly after birth; these include severe hypotonia, for example, presenting as a floppy baby, with weak fetal movements, arthrogryposis, polyhydramnios (due to poor fetal swallowing), pulmonary hypoplasia (due to reduced fetal respiratory movements), hydrops fetalis, and or stillborn. Occasionally, the baby’s clinical presentation has a 3- to 4-day latency. Other clinical manifestations include generalized weakness, facial diplegia, poor suck and feeding, a weak cry, intermittent cyanosis (especially during feeds), respiratory weakness and/or failure. Muscle stretch reflexes, sphincter function, and sensation are preserved. Ptosis and external ophthalmoplegia are paradoxically less frequent than in juvenile myasthenia gravis. The symptoms are transient, typically lasting about 3 weeks, but may extend for up to 3 months.101
When the mother has myasthenia gravis, diagnosis of TNMG is straightforward. Congenital myasthenia gravis syndromes, infantile botulism, congenital myotonic dystrophy, congenital muscular dystrophy, Moebius syndrome, and spinal muscular atrophy are in the differential diagnoses.102,103 Transient improvement following injection of 0.1 mg/kg of edrophonium supports a TNMG diagnosis.
After delivery, the maternal acetylcholine receptor antibodies are no longer pathogenic; breast milk contains the antibodies, but they do not appear to affect the infant.14There has not been any consensus as to breast feeding with transient neonatal myasthenia gravis; however, there is no evidence to suggest that breast feeding is contraindicated.
 TREATMENT
TREATMENT
Supportive treatment is necessary until the symptoms clear. If the infant is severely affected, ventilatory support and nasogastric feeding may be required. Acetylcholinesterase inhibitors such as pyridostigmine or neostigmine methylsulfate may be helpful.14 Intravenous immunoglobulin106,107 and exchange transfusion108 provide other therapeutic alternatives.
One recent transient neonatal myasthenia gravis study evaluated 73 mothers with myasthenia gravis who had 135 babies. Just one third of mothers had a neurologic evaluation during pregnancy. Transient neonatal myasthenia gravis children typically displayed signs of fetal distress during delivery (p = 0.05). The authors suggest that during pregnancy myasthenia gravis mothers benefit from a neurologic examination in collaboration with obstetricians. It is hoped that this would minimize risks and help decide the best delivery mode.110
OTHER NEUROMUSCULAR JUNCTION DISORDERS
 INFANTILE BOTULISM
INFANTILE BOTULISM
Infantile botulism is a relatively rare disorder typically seen in previously healthy infants between the third and sixth month of life.111 Infantile botulism particularly occurs in children residing in the middle Atlantic states, Utah, and California. Clostridium botulinum is an obligatory anaerobic, gram-positive spore-forming rod bacteria. As formula and or solid foods are introduced into the diet, the normal intestinal microflora change.112,113 This may enhance the ability of C botulinum to colonize the infant’s colon. This is not a contagious disease.112
Typically, the mother may first note that her baby is not “stooling” with normal frequency.114 Generalized muscle weakness develops. Poor feeding follows secondary to bulbar dysphagia, and poor suck impairs nursing. The face is typically expressionless, drooling is present, and a high-pitched, mewing cry frequently occurs.112 Aspiration often leads to hospitalization of an alert, afebrile, nonirritable infant with poorly reactive pupillary light reflexes, ophthalmoparesis, symmetric facial bulbar weakness, and generalized hypotonia.115,116
EMG demonstrates a significant incremental response (23–313%) to rapid repetitive stimulation (20 or 50 Hz).117(See eFig. 571.4  .) Both the botulinum toxin and the clostridium organism are recoverable from stool.118,119
.) Both the botulinum toxin and the clostridium organism are recoverable from stool.118,119
Treatment primarily is supportive. Intubation is often required acutely. Human-derived botulinum-immune globulin is beneficial when administered within the first 3 days.112,120,121 Antitoxin per se is not used for infantile botulism because of the possible anaphylactic risk. Early aggressive supportive care will ensure that these children all have an excellent outcome.
 CONGENITAL MYASTHENIA SYNDROMES
CONGENITAL MYASTHENIA SYNDROMES
Congenital myasthenia gravis syndromes comprise a group of widely differing, rare, familial neuromuscular transmission disorders, leading to fatigable weakness.120a These genetically determined “myasthenic” disorders usually present during the first years of life, but occasionally their onset is delayed. Clinically, ptosis and extraocular weakness are often more subtle than in juvenile myasthenia gravis. Additionally, bulbar, neck, and extremity weakness occur, sometimes with a restricted distribution. Respiratory distress leading to sudden death occurs rarely. EMGs have a decremental response similar to juvenile myasthenia gravis; this may be restricted to certain muscles and be present only intermittently. Testing for acetylcholine receptor antibodies is always negative. Pyridostigmine provides the primary treatment option.
 LAMBERT-EATON MYASTHENIC SYNDROME
LAMBERT-EATON MYASTHENIC SYNDROME
Although Lambert-Eaton myasthenic syndrome is often not considered in the differential diagnosis of pediatric proximal weakness, it occasionally occurs during the primary school years.122-125 Sometimes the clinical presentation may emulate myasthenia gravis, as a number of Lambert-Eaton myasthenic syndrome patients have ocular/bulbar symptoms and signs.126 Often there is an associated pandysautonomia with decreased salivation, dry eyes, and sluggish pupillary responses.
Diagnosis depends on two modalities. EMG demonstrates a classic 100% (doubling) facilitation with repetitive motor nerve stimulation. (See eFig. 571.5.  ) Serum antibody testing for the P/Q 65 voltage-gated calcium channel antibody is the definitive diagnostic stool. Treatment options include pyridostigmine, prednisone, and steroid-sparing medications.
) Serum antibody testing for the P/Q 65 voltage-gated calcium channel antibody is the definitive diagnostic stool. Treatment options include pyridostigmine, prednisone, and steroid-sparing medications.
REFERENCE
See references on DVD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


