Reference
Classification system
Epstein [2]
Intrinsic
Diffuse
Focal
Cervicomedullary
Exophytic
Anterolateral
Posterolateral
Disseminated
Positive CSF cytology
Positive myelography
Choux et al. [3]
Type I
Diffuse
Type II
Intrinsic, focal
Type III
Exophytic, focal
Type IV
Cervicomedullary
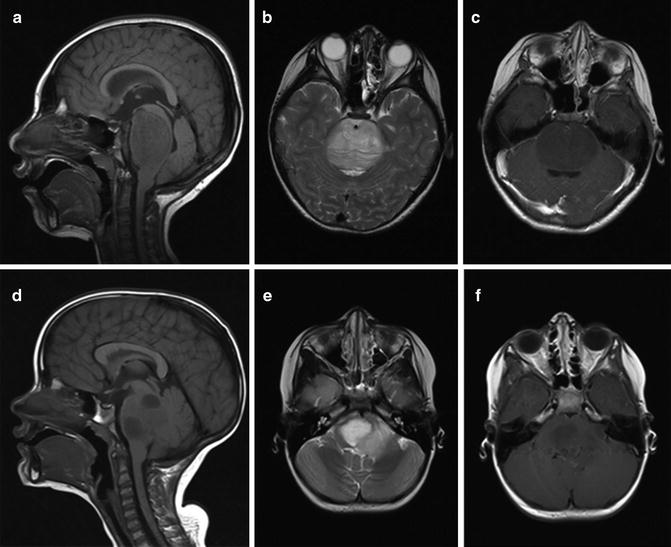
Fig. 11.1
(a–c) A 7-year-old male with DIPG. (a) Sagittal T1 with gadolinium, (b) axial T2, and (c) axial T1 with gadolinium. (d–f) A 6-year-old female with DIPG. (a) Sagittal T1 with gadolinium, (b) axial T2, and (c) axial T1 with gadolinium. Both tumors demonstrate minimal contrast enhancement
Clinical Features
The clinical features of patients with brainstem tumors vary with respect to tumor location and the nature/pattern of growth of these tumors. Patients with DIPG have a short latency (usually less than 2–3 months) between the onset of clinical symptoms and diagnosis. The classic triad of symptoms includes cerebellar signs (e.g., ataxia, dysmetria, dysarthria), long–tract signs (e.g., increased tone, hyperreflexia, clonus, Babinski sign, motor deficit, etc.), and isolated or multiple cranial nerve palsies (unilateral or bilateral), more commonly 6th and 7th nerve palsies. Patients with diffuse brainstem tumors associated with neurofibromatosis type 1(NF1) usually have tumor that may mimic DIPG on imaging. However, in the context of NF1, these are low-grade gliomas (LGG, WHO grades I–II) that can be asymptomatic or diagnosed in the context of an insidious history of isolated cranial nerve palsy or motor deficit. Careful clinical examination for the stigmata of NF-1 and family history should help in the identification of these lesions that usually do not require any active treatment. In DIPG, signs and symptoms of increased intracranial pressure (due to obstructive hydrocephalus from expansion of the pons) are seen in less than 10 % of children. Various other nonspecific symptoms present either at the time of diagnosis or at the time of progression include sensory abnormalities, behavioral changes (night terrors, pathologic laughter, and separation anxiety), urinary problems, declining school performance, respiratory symptoms including sleep apneas, etc.
Diagnostic Evaluation
Neuroimaging
The two most commonly used neuroimaging modalities for diagnosing DIPG are CT and MRI. DIPGs appear hypodense on CT with a nonspecific pattern of enhancement and comprise almost 80 % of all pontine tumors. Invasion into the adjacent midbrain and medulla is common, but they rarely affect the fourth ventricle (in 10 % of cases). The diagnosis of DIPG is mostly based on characteristic MRI findings in the presence of typical clinical features (Fig. 11.1) [4]. The classic MRI findings include the appearance of a diffusely expansile pontine mass with or without encirclement of the basilar artery. Most diffuse brainstem tumors appear to be hypointense on T1-weighted MR images and hyperintense on T2-weighted images. Prominent peritumoral edema is commonly seen. The ventral pons may appear swollen and infiltrated. There is variable contrast enhancement ranging from homogeneous rim enhancement to patchy enhancement to the complete absence of enhancement.
Other MR-based imaging technologies can play a very important role in the management of DIPG, especially in monitoring of response to therapy. Response assessment during or post chemotherapy/targeted therapy by MRI is done using the Response Assessment in Neuro-Oncology (RANO) criteria [5] with decreased tumor size, decreased steroid use, and improved neurologic symptoms indicative of response. These criteria were developed primarily for contrast-enhancing tumors like supratentorial GBM in adults (WHO grade IV), and DIPGs frequently do not enhance or they may exhibit variable patterns of enhancement. Conventional MRI instructs us regarding tumor structure and location and cannot reliably differentiate therapy-related phenomena such as efficacy, pseudo-progression, or pseudo-response. Hipp et al. [6] used multiparametric imaging in a prospective study to evaluate outcome of children with DIPG. In their study, increased perfusion (as determined using DSC MRI) at any single time point was associated with shorter survival (RR = 4.91), and increasing perfusion over time was a poor prognostic factor. In other studies using MR spectroscopy (MRS) in DIPG, the choline:N-acetylaspartate ratio (CHO:NAA) has been shown to be prognostic, with those patients having a CHO:NAA ratio higher than the median of 2.1 demonstrating a greater risk of early mortality compared to patients with CHO:NAA ≤2.1. Changes in this ratio during follow-up had an impact on prognosis; increase in the CHO:NAA ratio was inversely associated with survival, while a decreasing CHO:NAA ratio was associated with a longer life expectancy. Neuroimaging using MRS can also be used in DIPG to evaluate the pyramidal tracts when combined with diffusion tensor (DT) tractography to differentiate between benign and malignant lesions (such as brainstem LGG associated with NF-1) and selection of a tumor site for biopsy to differentiate from nonneoplastic processes including neurodegenerative conditions (Alexander disease, ADEM/acute demyelinating encephalomyelitis, central pontine myelinolysis), infections (paracoccidioidomycosis), etc.
Pathology
Role of Biopsy
Historically, prior to CT and MRI, stereotactic brainstem biopsies were performed on a regular basis for histological confirmation of the diagnosis. Due to the heterogeneity of these tumors, the significant morbidity potentially associated with the biopsies, the minimal alteration of the treatment plan based on biopsy results, the prevalence of poor candidates for biopsy at the time of presentation (i.e., those with focal neurological deficits, increased ICP), and the widespread availability of MRI with characteristic radiologic findings, routine biopsy as the standard of care was discontinued in the early 1990s [7].
However, compared to the majority of centers in North America, routine biopsy of children with suspected DIPG has been performed in Europe in the last decade (UK and France). In two of the largest series of brainstem tumor biopsies in children [8, 9], no mortality was reported and transient reversible morbidity (cranial nerve palsy, worsening hemiparesis) lasting only a few days were reported in less than 10 % of patients. More recently, Grill et al. reported on 20 newly diagnosed DIPG patients who had a stereotactic biopsy [10] and new multidisciplinary consensus statements have been developed to gliomas, including DIPG [11]. In summary, the authors conclude that brainstem biopsy is relatively safe in experienced hands using modern neurosurgical techniques. The rationale behind this paradigm shift is that stereotactic biopsies may enable to perform prognostic genomic testing on small tissue samples (such as H3.3 mutations with increased or decreased survival) and allow identification of potentially druggable targets. With the availability of small molecule inhibitors to these targets, it may be possible to develop new clinical approaches. However, this approach still remains experimental, and pilot studies in Europe and North America are ongoing to confirm the safety of stereotactic biopsies in the context of multicenter trials and their potential for influencing therapy.
Histopathology
The histologic appearance of diffuse intrinsic pontine gliomas is similar to that of other high-grade astrocytomas in other locations (grades III–IV). Classic features including an increased mitotic index, cellular anaplasia, areas of tumor necrosis, and hemorrhage with increased angiogenesis are most often seen. Pretreatment biopsy samples correlate well histologically with autopsy samples. The utility of differentiating between WHO grade III and IV histology in the pons is probably not relevant due to the uniformly poor outcome in all these patients. Biopsies when indicated (atypical radiologic features, exophytic tumors, long clinical latency) are obtained from areas of tumors easily accessible to the neurosurgeon, and due to tumor heterogeneity might not be truly representative (i.e., showing features of lower-grade tumor such as grade II). It has been shown that tumor grade by histology is not predictive of prognosis in typical DIPG. DIPG predominantly spread by contiguity extending rostrally into the brainstem and caudally into the cerebellar peduncles and the medulla. Leptomeningeal dissemination is seen in 10–20 % of the cases at the time of diagnosis. However, this number increases to 50–60 % in patients with recurrent or progressive DIPG or at the time of autopsy. For this reason, some authors consider that baseline MRI of both the brain and spine at diagnosis followed by periodic surveillance of the spine during therapy should be performed.
Molecular Genetics and Tumor Biology
A number of recent comprehensive studies have established that DIPGs can be considered a separate biologic entity when compared to adult and the majority of pediatric supratentorial GBMs at the genetic and epigenetic levels. The recently proposed integrated genomic classification categorizes the majority of DIPG into the histone variant H3.3 K27 group (midline infratentorial tumors with the H3.3 K27M mutation).
Recurrent somatic driver mutations in the H3F3A gene that affect its coded protein, the replication-independent histone 3 variant (H3.3), lead to an amino acid substitution at a key residue, namely, lysine (K) 27. This K27M mutation is seen in over 70 % of DIPG cases [12]. The K27M mutations are also seen in 10–20 % of the supratentorial midline HGGs. This mutation is seen in both pretreatment biopsy samples and autopsy material indicating that it is present at diagnosis and not induced by therapy. DIPG can be classified into wild type and mutated based on the K27M-H3.3 mutation with distinct clinical, genetic, and prognostic features [12]. Tumors with a wild-type (wt) H3.3 at the K27 residue often have atypical clinical presentation (long latency of symptoms, atypical MRI findings) and show high-grade histology on biopsy samples with a few long-term survivors when compared to the H3.3 K27M mutated DIPGs. Also specific focal chromosome number gains involving the ASAP2 (2p25.1) and MYCN (2p24.3) genes may be seen in K27 wild-type tumors [12]. The H3.3 K27M-mutated DIPGs have worse overall survival when compared to wild-type tumors, and this association is independent of patient age and histological diagnosis [12]. These mutated tumors have glial differentiation on histology and are exclusively associated with gain or amplification of PDGFRA (4q12, seen in 40 % of cases) and MYC/PVT1 (8q24). In one study, TP53 mutations were seen in 77 % of DIPG tumors and were associated with both wild-type and mutated H3.3 tumors [12]. ATRX mutations are infrequent in DIPGs (9 %) when compared to supratentorial pediatric HGGs (29 %) and are seen in mutated K27M-H3.3 tumors in children of an older age group [12]. In another study, mutations involving the replication-dependent histone 3 variant (H3.1) that lead to the same amino acid substitution (H3.1 K27M) were seen in 9 % of the patients with DIPG [13]. A summary of the DIPG classification based on the H3.3 mutation status is given in Table 11.2.
Table 11.2
Integrated genomic classification of DIPG
H3.3 or H3.1 (K27M mutant) | H3.3 or H3.1 (wild type) | ||
|---|---|---|---|
Clinical features | |||
Age in years (mean) | 8.1 | 4.6 | |
Atypical clinical features (longer duration of symptoms, atypical radiology) | − | + | |
Overall survival | < 1 year | 4–5 years; some long-term survivors | |
Genomic features | |||
Copy number Abnormalities (gains/amplifications) | 1. PDGFRA (4q12) | 1. ASAP2 (2p25) | |
2. MYC/PVT1 (8q24) | 2. MYCN (2p24) | ||
TP53 mutation | ++ (~80 %) | ++ (~80 %) | |
ATRX | + (older children) | – | |
Potential therapeutic targets/agents | |||
Histone modifying agents + RTK Inhibitors | |||
When compared to pediatric high-grade gliomas (pHGG), DIPGs show a higher frequency of chromosomal imbalance with gains of chromosomes 2, 8q, and 9q. Losses of chromosomes 16q, 17p, and 20p occur more frequently in DIPG when compared to adult HGG [14]. Receptor tyrosine kinases (RTKs) appear to be upregulated at the genomic or expression level (or both) in the majority of pediatric DIPGs. The most common recurrent focal gain in pediatric DIPG encompasses PDGFRA, with amplification in at least 30 % of DIPGs, and an even larger number showing overexpression at the RNA and protein levels. EGFR gain does not appear to be a frequent event in pediatric DIPG. Multiple dual amplifications of other RTKs like MET, IGF1R, and ERBB4 alone or in combination with PDGFRA are also reported in DIPG. These discoveries, along with reported oncogenic PI3K pathway mutations in PI3KCA, support new treatment approaches targeting this pathway [10, 14–16].
Cell of Origin and Animal Models
There are several unique features of DIPG, which suggest that these tumors are related to dysregulation of a normal postnatal neurodevelopmental process (reviewed in [14]): distinct age of incidence (5–10 years of age), specific anatomical site, and unique biological features (H3.3 K27M mutations, RTK and cell cycle regulatory pathways which play a role in normal brainstem embryogenesis). Monje et al. identified a neural precursor-like cell population in the human ventral pons that is linked (both anatomically and temporally) to the incidence of DIPG [17]. During normal growth and development of the brain, these pontine precursor-like cells (PPC) are spatiotemporally restricted to the ventral brainstem, and the density of the PPC varies during different time periods (present during infancy, decreases by 2 years of age, and increases again during middle childhood). Using mouse models, the authors showed that the signaling by the Hedgehog pathway is increased and contributes to PPC proliferation. This time period in the mouse corresponds to the middle childhood period in humans when DIPG occurs. However, aberrant Hedgehog signaling alone did not result in DIPG in this mouse model, indicating the need for additional genetic mutations (PDGFRA, MYC/PVT1, TP53, etc.) [17].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


