Diagnostic Testing in Rheumatology
Peter A. Nigrovic
Laboratory and imaging studies rarely establish a definitive diagnosis of rheumatologic diseases in children. The prevalence of rheumatic diseases in childhood is low, and even very specific tests generate high false-positive rates unless there is a well-founded clinical suspicion for a particular illness, based upon an informed differential diagnosis guided by the history and physical exam. The utility of an assay will vary with the nature and stage of the illness. They can provide information that is useful for the evaluation and treatment.
LABORATORY STUDIES
MARKERS OF INFLAMMATION
Most rheumatologic diseases arise out of aberrant immune attack on normal cells and tissues. In many cases this immune activity is reflected in indices of systemic inflammation. Two principal markers are the erythrocyte sedimentation rate (ESR) and the C-reactive protein (CRP), but other tests also may be useful. It is important to remember that localized inflammation, such as glomerulonephritis in systemic lupus erythematosus (SLE), or synovitis in pauciarticular juvenile idiopathic arthritis (JIA), may not be reflected in these indices.
 ERYTHROCYTE SEDIMENTATION RATE
ERYTHROCYTE SEDIMENTATION RATE
The ESR measures the speed with which red blood cells precipitate out of suspension in a sample of anticoagulated blood, and is quantitated at mm/hour. In the patient with inflammation, hepatic synthesis of positively charged, acute phase reactants such as fibrinogen enables negatively charged erythrocytes to overcome electrostatic repulsion and stack together in columns (rouleaux), which fall out of suspension in the blood more swiftly. The ESR thus serves as an indirect measure of the hepatic acute phase response.1 Changes in the ESR follow a time course commensurate with hepatic protein synthesis and the degradation and clearance of the relevant proteins, rising and falling gradually over the course of days.
The advantages of the ESR are its technical simplicity and low cost, and its long track record of use. While mild elevations of ESR (<40 mm/h, for example) arise in the course of many routine illnesses, more substantial elevations warrant scrutiny for a worrisome underlying cause such as infection, malignancy, or rheumatologic disease.2
Although it is a robust and useful tool, the ESR has limitations intrinsic to the assay. Processes that alter red cell properties or the plasma protein milieu may alter the ESR. ESR decreases with elevated hematocrit, aberrant red cell shape (eg, sickle cell disease and spherocytosis), and disseminated intravascular coagulation (DIC) due to consumption of circulating fibrinogen. ESR rises with anemia, pregnancy, nephrotic syndrome, and hypergammaglobulinemia, where positively charged immunoglobulins promote rouleaux formation that can markedly elevate ESR in the absence of systemic inflammation.
 C-REACTIVE PROTEIN
C-REACTIVE PROTEIN
CRP is an innate antibacterial compound that is produced as part of the hepatic acute phase response. Present at low to undetectable levels in health, the CRP rises briskly within hours of the onset of inflammation, and once an inflammatory stimulus ceases the CRP level falls with a half-life of 19 hours.4 Accordingly, the CRP reflects hepatic acute phase protein synthesis more tightly and directly than does the ESR and it is not influenced by the factors that confound the ESR, such as sickle cell disease.5,6 However, certain conditions manifest with CRP values lower than might be expected, including SLE.7 In practice, both CRP and ESR shed light on the global inflammatory state but vary in their reliability from patient to patient. Often, interpreting the two together provides the most useful information.
 OTHER INFLAMMATORY MARKERS
OTHER INFLAMMATORY MARKERS
Beyond the ESR and CRP, other blood indices can reflect systemic inflammation, including the white blood cell count, the platelet count, ferritin, the von Willebrand factor antigen, and total immunoglobulin level. The importance of each of these as a disease marker varies with the condition in question. 
AUTOANTIBODIES IN THE RHEUMATIC DISEASES
The production of antibodies to self-antigens is a hallmark of many rheumatologic diseases (Table 200-1). The presence of these antibodies indicates a defect in normal mechanisms of immune tolerance. Many of these antibodies serve simply as diagnostic markers, whereas others may participate directly in disease pathogenesis. Because autoantibody titers generally do not correlate with disease activity, in most cases serial monitoring of autoantibody levels contributes little to disease management.
 RHEUMATOID FACTOR (RF)
RHEUMATOID FACTOR (RF)
Rheumatoid factor is the general term given to antibodies that recognize the Fc portion of IgG.
Rheumatoid factor may be observed in many states of immune activation, including multiple autoimmune diseases, chronic bacterial infection, and hepatitis C or cryoglobulinemia (Table 200-1). The prevalence of a positive RF in normal children is less than 3%, and then typically in low titer.10-12 Among the childhood rheumatic diseases, the presence of rheumatoid factor defines a subset of polyarticular JIA, and is associated with a chronic, aggressive course with little likelihood of permanent disease remission13-15 (see Chapter 201). RF is also observed in Sjögren syndrome, often at high titer. 
Table 200-1. Important Autoantibodies in the Rheumatologic Diseases
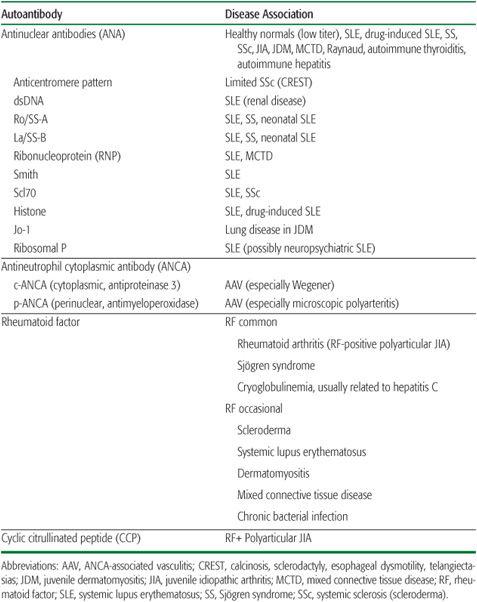
The diagnostic role for rheumatoid factor testing in pediatrics is limited. While the presence of RF in the setting of an active polyarthritis helps to classify and prognosticate, it does not contribute to making a definitive diagnosis of JIA.19 Since RF is rarely meaningfully positive in arthritis occurring in younger children (eg, before age 8), it is not routinely warranted as part of the evaluation of arthritis below this age. Even in adolescents, the likelihood that a positive rheumatoid factor will be clinically useful is small; in one study, more than half of positive RF tests among patients referred to a pediatric rheumatology clinic were considered to be false positives. A high titer RF in a suggestive clinical setting supports the diagnosis of Sjögren syndrome. The optimal evaluation for a child noted to have a high-titer rheumatoid factor in the absence of evident clinical disease remains undefined. RF is known to predate the onset of arthritis in a substantial fraction of adult patients who go on to have RA.20 Other diagnoses to consider include Sjögren syndrome, cryoglobulinemia, and chronic bacterial infection.
 ANTICYCLIC CITRULLINATED PEPTIDE ANTIBODIES
ANTICYCLIC CITRULLINATED PEPTIDE ANTIBODIES
The identification of antibodies against citrullinated peptides represents a recent advance in the understanding of the pathogenesis of some forms of arthritis.21 Citrulline is an amino acid that results from the enzymatic deimination of arginine. The presence of autoantibodies against citrullinated peptides is highly specific for adult RA and its juvenile counterpart, RF-positive polyarticular JIA, and is generally assessed as reactivity to a synthetic cyclic citrullinated peptide (CCP). CCP-positive arthritis overlaps substantially with RF-positive arthritis, with the advantage of fewer false positive results than are seen with RF.11,22 Like RF, CCP positivity is associated with more aggressive arthritis unlikely to enter spontaneous remission. Its utility lies primarily in helping to classify and stratify risk levels of arthritis rather than confirming the diagnosis.
 ANTINUCLEAR ANTIBODIES (ANA)
ANTINUCLEAR ANTIBODIES (ANA)
The most common autoantibodies detected in children with rheumatic disease are the antinuclear antibodies (ANA). ANA is assessed by layering the patient’s serum over cells fixed to a surface, counterstaining with a fluorescent anti-human immunoglobulin, and visually assessing the lowest serum dilution at which fluorescence remains detectable. Normal is less than 1:40 in most laboratories. The significance of a positive test tends to rise with antibody titer. Further specificity is provided by testing for antibodies against defined targets, sometimes termed extractable nuclear antigens (ENAs), by enzyme-linked immunosorbent assay. The immunofluorescence pattern of staining (for example, homogenous, speckled, rim, nucleolar) is of limited utility now that specific antigen testing is available. The single exception is the anticentromere pattern observed in limited systemic sclerosis (see Chapter 206).
ANAs are found in a wide range of immune disorders in children. These include SLE and related conditions, JIA, and autoimmune hepatitis. ANAs are also found at low titer in many normal children, ranging from 1.6% to 15% in healthy children tested at a serum dilution of 1:10.24-26 Given the low prevalence of ANA-associated disorders, the ANA has a poor positive predictive value in the absence of compelling historical, examination, or laboratory features of a specific rheumatic illness.27,28 In particular, SLE is highly unlikely in children with an ANA titer of 1:160 or less.27 By contrast, the presence of an ANA at any titer in a patient with JIA is relevant as a predictor of elevated uveitis risk (see Chapter 201). Follow-up of patients referred to pediatric rheumatology clinic with a positive ANA but no clinical rheumatic disease at presentation has established that subsequent development of an ANA-related disease is rare, although the positive ANA itself may persist for years.26,29
The clinical significance of a positive ANA may be defined further by testing for specific, disease-associated targets of the autoantibodies.31,32 These specific antibodies are rare in ANA-positive healthy children and those with JIA, but they are usually positive in patients with SLE and related disorders (Table 200-1). Most patients with SLE have at least one of these autoantibodies; dsDNA and Sm are relatively specific for this diagnosis. Within SLE, the presence of dsDNA correlates with an increased risk for nephritis, and the titer tends to rise and fall with disease activity.33 Ro and La are found in SLE but are prominent in Sjögren syndrome. Maternal Ro and La antibodies are associated with a risk for neonatal SLE in the newborn, in particular congenital heart block, because of cross-reactivity with antigens expressed in the fetal myocardium.34 RNP is common in SLE generally but is found in high titer in mixed connective tissue disease (MCTD), an overlap syndrome with features of SLE, scleroderma, and dermatomyositis. Scl70 is associated with SLE and scleroderma. Antihistone antibodies are common in patients with SLE, but they are a hallmark of SLE induced by exposure to drugs such as hydralazine and procainamide when found in the absence of antibodies against other ENAs.35 However, SLE induced by other drugs, including minocycline, is commonly negative for antihistone antibodies, so a negative test does not definitively rule out the possibility of drug-induced SLE. Jo-1 positivity correlates with increased risk of interstitial lung disease in patients with dermatomyositis. Antiribosomal P antibody has been associated with CNS manifestations of SLE but is of limited sensitivity and specificity.
 ANTIPHOSPHOLIPID ANTIBODIES
ANTIPHOSPHOLIPID ANTIBODIES
The term antiphospholipid antibodies (aPLs) refers broadly to a group of autoantibodies associated with increased clotting risk in some patients. They are believed to promote coagulation by interference with normal anticoagulant mechanisms and by the direct promotion of inflammation.
The most commonly measured aPLs are anticardiolipin antibodies and the lupus anticoagulant. Testing can also be performed to look for antibodies against specific PL-associated proteins, including beta-2 glycoprotein 1 (β2GP1) and prothrombin.
The clinical significance of a positive test for aPL antibodies changes dramatically with context. Transient aPLs appear to be rather common after viral infections and from other triggers.36 In past years these were usually uncovered during routine testing for syphilis (the VDRL test employs cardiolipin as antigen) and during preoperative testing of the partial thromboplastin time (PTT), which is prolonged by a lupus anticoagulant. Follow-up of such patients has generally been reassuring, with a low risk of pathologic thrombosis.37 By contrast, an aPL identified in a patient with a history of clot is likely of pathogenic importance and can help to explain the index event.
There is no role for aPL testing as a screen of asymptomatic patients. In patients with SLE, testing for anticardiolipin and lupus anticoagulant may help define thrombosis risk. By contrast, in a patient with unexplained thrombosis, testing for anticardiolipin, lupus anticoagulant, and often anti-β2GP1 is critical to define the clotting diathesis; even more refined testing may be appropriate if the clinical suspicion warrants. The role of antibody titer monitoring to assess future risk in patients who have experienced aPL-related thrombosis is controversial. Even if antibody levels fall below the limit of detection, the pathogenic plasma cell clone remains and could be reactivated by a viral illness or other unpredictable event to again produce pathogenic antibodies. Thus, these patients may continue to be at increased risk of pathologic thrombosis indefinitely.
 ANTINEUTROPHIL CYTOPLASMIC ANTIBODIES
ANTINEUTROPHIL CYTOPLASMIC ANTIBODIES
Antineutrophil cytoplasmic antibodies (ANCAs) were originally identified in patients with Wegener granulomatosus as an antibody-mediated reactivity to the cytoplasm of fixed neutrophils.39,40 Subsequently two staining patterns were identified: a uniform cytoplasmic stain (c-ANCA) and a stain clustered around the borders of the nucleus (perinuclear or p-ANCA). In patients with ANCA-associated vasculitis (AAV), these patterns correspond to antibody reactivity against specific antigens: antiproteinase 3 (PR3) for c-ANCA and antimyeloperoxidase (MPO) for p-ANCA. ELISA testing to confirm that a p-ANCA targets MPO is important because of the relative nonspecificity of this immunofluorescence pattern.
ANCA positivity defines a class of life-threatening, small-vessel vasculitides affecting the renal and pulmonary vessels, among others (see Chapter 203). PR3-ANCA (c-ANCA) occurs most often in Wegener granulomatosis (WG), a granulomatous vasculitis affecting the sinuses, airway, lungs, and kidneys. MPO-ANCA (p-ANCA) associates more tightly with microscopic polyangiitis (MPA), a vasculitis associated with pulmonary hemorrhage and less prominent renal disease. ANCA positivity also may be seen in Churg-Strauss vasculitis, but the association in this condition is less consistent. In the proper clinical setting, the specificity of a positive ANCA assay is very high, in some cases obviating the need for tissue biopsy. However, in situations in which the clinical picture diverges from typical findings in a small-vessel vasculitis, false positives may be observed, especially for p-ANCA. Conversely, cases of so-called “limited Wegener” (sinus and upper respiratory disease sparing kidney and lung parenchyma) are often ANCA negative. The pathogenic role of ANCA remains unclear.
OTHER TESTS IN RHEUMATOLOGY
 JOINT FLUID EXAMINATION
JOINT FLUID EXAMINATION
Inflamed joint tissues produce an excess of synovial fluid that can be sampled clinically by joint aspiration. Normal joint fluid is straw colored, relatively viscous, and contains no more than a few hundred white blood cells/ml, including few if any neutrophils. Joint inflammation of any cause results in the influx of inflammatory cells, most commonly neutrophils. In children, the major purpose of joint aspiration is to exclude bacterial infection; joint fluid examination under polarized light microscopy is also the method of choice to evaluate for crystal-induced synovitis (gout, pseudogout). An elevated joint fluid white blood cell count (eg, >50,000/ml) with a neutrophil predominance is usual in bacterial infections, but this degree of pleocytosis also may be seen in other joint processes, such as JIA or Lyme disease. Thus, infection cannot be confirmed without consistent findings on Gram stain and culture. When bacterial infection is not suspected, the characterization of joint fluid contributes minimally to the diagnostic evaluation of pediatric arthritis. The presence of grossly bloody or rust-colored joint fluid is observed with intra-articular trauma, hemophilia-associated arthropathy, and, more rarely, synovial neoplasms such as pigmented villonodular synovitis (PVNS) and synovial sarcoma. Joint fluid may also be tested for B. burgdorferi DNA by polymerase chain reaction (PCR), but this test is imperfectly sensitive, especially after treatment with antibiotics.41 Assays for other joint fluid parameters such as viscosity, glucose or LDH are rarely helpful.42
 HLA TYPING
HLA TYPING
The human leukocyte antigen (HLA) locus on chromosome 6 encodes the critical antigen-presenting molecules major histocompatibility complex (MHC) I and II. These receptors are variable across the human population and help determine the antigens to which an individual may develop an immune response. Many autoimmune diseases exhibit strong genetic associations with particular HLA alleles. Among the strongest are the association of the MHC I molecule HLA-B27 with ankylosing spondylitis (relative risk ≈ 20) and of specific HLA-DR alleles (MHC II) with arthritis exhibiting RF and/or anti-CCP antibodies (relative risk ≈ 5–11).43-45 While these associations are pathophysiologically significant, the value of clinical testing for HLA type is controversial. Positive and negative predictive values for individual patients are both limited. The major clinical utility of HLA-B27 status is in cases where a diagnosis of spondyloarthropathy remains uncertain after clinical and imaging evaluations have been performed.
 COMPLEMENT
COMPLEMENT
The complement system is discussed in Chapter 189. In clinical practice, quantitation of the integrity and activity of the complement system can provide important diagnostic information. Most commonly measured are the levels of C3 and C4. Low levels of these proteins usually reflect complement consumption by an inflammatory process, although increased hepatic synthesis of both these acute phase reactants may mask activation of the complement system. Both C3 and C4 are typically low in patients with active SLE, though they do not invariably normalize with successful treatment. C3 may be low in patients with post-streptococcal glomerulonephritis, whereas C4 is typically depressed (sometimes with C3 also) in cryoglobulinemia.
 CRYOGLOBULINS
CRYOGLOBULINS
Cryoglobulins are antibody–antibody or antibody–antigen complexes that remain soluble at physiologic temperature but precipitate in the cold. They are associated with superficial and sometimes deep small-vessel vasculitis resulting in features such as cutaneous ulcers and mononeuritis multiplex (see Chapter 203). Cryoglobulins may be monoclonal (Type I), arising as part of a clonal B cell dyscrasia, or polyclonal, involving multiple different antibodies. Polyclonal (mixed) cryoglobulins are far more common, and are sometimes divided into subtypes (Type II and Type III) depending on whether the associated antiantibody (rheumatoid factor) activity is monoclonal or polyclonal, a distinction of little practical importance. All types of cryoglobulins are rare in children.
The measurement of cryoglobulins requires that the blood sample remain warm until the serum can be removed, to prevent loss of the cryo-precipitate in the cellular pellet. Blood should be drawn into a warmed, anticoagulant-free tube, kept warm in transport, and spun down in a warmed centrifuge, after which the serum is placed in a refrigerator for several days to observe for precipitate formation. The degree of cryoglobulinemia is quantitated as a fraction of serum volume taken up by the precipitate (cryocrit), and can be followed to assess response to therapy. The leading cause of mixed cryoglobulinemia is chronic hepatitis C infection, so an anti–hepatitis C antibody level should be checked when cryoglobulinemic vasculitis is a strong consideration. Rarely the hepatitis C antibody may be completely bound up in the cryoglobulins and will be falsely negative, although a hepatitis C RNA assay will still be positive.
IMAGING STUDIES
Imaging is an important adjunct to clinical and laboratory evaluation in the rheumatic diseases. Broadly speaking, plain radiography and ultrasound have low risk and cost but limited sensitivity. Computerized tomography (CT) provides excellent spatial resolution of bony structures but limited soft tissue characterization. Magnetic resonance imaging (MRI) offers the greatest visualization of soft tissue structures, especially cartilage and synovium. Unfortunately, this must be balanced against high cost and the need for sedation in younger children. The choice of imaging modality depends on the clinical scenario, and in all cases it must be remembered that radiographic studies are seldom diagnostic, but rather are useful for confirming or refuting the clinical impression.
 DISEASES OF THE JOINTS
DISEASES OF THE JOINTS
Many of the hallmark rheumatic diseases primarily affect the musculoskeletal system. Imaging can help define the extent of joint inflammation or injury. In arthritis, the synovial lining hypertrophies and generates an abundance of inflammatory joint fluid. Over time, this process erodes cartilage and bone, damages ligaments and tendons, and induces regional osteopenia through inflammation and disuse. In skeletally immature children, inflammation also impacts surrounding growth centers and can affect bone growth and morphology. These processes can be assessed and followed by imaging.48
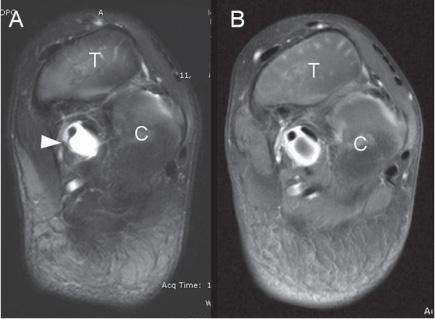
In the acutely inflamed joint, plain films can document the presence of thickened soft tissue and effusion (eFig. 200.1  ). Radiographs can assist with the initial evaluation if the differential diagnosis includes such entities as trauma, tumor, or chronic osteomyelitis; otherwise, clinical examination without imaging is typically sufficient. In deeper joints that cannot be palpated directly, particularly the hip, ultrasound can identify swelling and guide aspiration if necessary. If the joint examination for active synovitis is equivocal, MRI can generally provide a definitive answer. MRI reliably detects even small effusions, and the intravenous contrast agent gadolinium highlights the inflamed synovial lining (Fig. 200-1). This is of particular utility for joints that are difficult to examine, such as the sacroiliac joints (eFig. 200.2
). Radiographs can assist with the initial evaluation if the differential diagnosis includes such entities as trauma, tumor, or chronic osteomyelitis; otherwise, clinical examination without imaging is typically sufficient. In deeper joints that cannot be palpated directly, particularly the hip, ultrasound can identify swelling and guide aspiration if necessary. If the joint examination for active synovitis is equivocal, MRI can generally provide a definitive answer. MRI reliably detects even small effusions, and the intravenous contrast agent gadolinium highlights the inflamed synovial lining (Fig. 200-1). This is of particular utility for joints that are difficult to examine, such as the sacroiliac joints (eFig. 200.2  ). MRI can identify early erosions of cartilage and bone that remain undetected by plain film.
). MRI can identify early erosions of cartilage and bone that remain undetected by plain film.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


