Neonatal Jaundice and Hemolytic Disease of the Newborn
INTRODUCTION
Neonatal jaundice, implying a yellow color of the skin and mucous membranes due to bilirubin deposition, occurs in about 60% of infants and even more often in those exclusively breast feeding. In most cases, the jaundice will be mild or moderate, and the total serum bilirubin (TSB) will not endanger the newborn infant. Occasionally, the TSB may increase to potentially dangerous levels and require treatment with phototherapy or rarely exchange transfusion to offset any further increase. Rarely, the TSB may rise to hazardous levels, at which bilirubin may cross the blood-brain barrier and enter the brain cells. The result will be the complication of acute bilirubin encephalopathy (ABE), in many cases with the devastating chronic sequela of choreoathetotic cerebral palsy known as kernicterus, or death. Despite attempts to eliminate this condition, kernicterus, although a rare condition, is still with us and accompanies us into the third millennium. Many cases should be preventable. Because the implications for affected individuals are lifelong, the public health aspects are necessarily of major importance. Understanding of the metabolism of bilirubin and the pathophysiology of hyperbilirubinemia are crucial to the prevention of this condition, as discussed in this chapter.
EPIDEMIOLOGY
Was There a Disappearance and Resurgence of Kernicterus?
Some authors refer to a disappearance and then resurgence of reported cases of kernicterus in Westernized countries during the last decades.1–4 Others opine that the condition never completely disappeared and is still being seen, both in countries with advanced medical systems and in developing countries.5 There can be no doubt that widespread use of the 2 mainstays of treatment of hyperbilirubinemia, exchange transfusion and phototherapy, along with immune prophylaxis of Rh isoimmunization, have prevented many cases of extreme hyperbilirubinemia. If the condition did indeed disappear, however, there can be no explanation for the cases of kernicterus still occurring due to conditions including glucose-6-phosphate dehydrogenase (G-6-PD) deficiency and direct antiglobulin titer (DAT) positive ABO isoimmunization, conditions that, in themselves, did not disappear.6,7
In favor of the resurgence theory, following a period during which few cases of kernicterus were reported, several case reports were published from the United States,8–10 followed by reports of the US-based Kernicterus Registry.7,11 In Denmark, Ebbesen12 did find cases of bilirubin encephalopathy between 1994 and 2001, whereas in a nationwide search, he uncovered no cases during the 20 years preceding 1994.
On the other hand, in the United States, Burke et al reported a 70% decrease in the number of hospitalizations between 1988 and 2005 of neonates diagnosed with kernicterus.13 Brooks et al found a constant incidence of kernicterus in California occurring during 2 time epochs: 1988–1993 and 1994–1997.14 On a national US basis, mortality data due to kernicterus from the Centers for Disease Control and Prevention databases revealed 31 kernicterus-related deaths between 1979 and 2006, the mortality during the first 14 years of the study period being similar to that of the second.13
The debate whether kernicterus did or did not disappear and then reappear is overshadowed by that fact that, well into the third millennium, cases of kernicterus are still being reported with major public health effects on neonatal and childhood morbidity and mortality.
How Frequent Is Kernicterus Currently?
National Statistics
Kernicterus is not a reportable disease, and the exact incidence may not be able to be accurately determined. From the data available, it appears that the incidence varies widely from country to country, as seen in Table 30-1.
Table 30-1 Incidence, per Live Births, of Acute Bilirubin Encephalopathy and Kernicterus in Westernized Countriesa
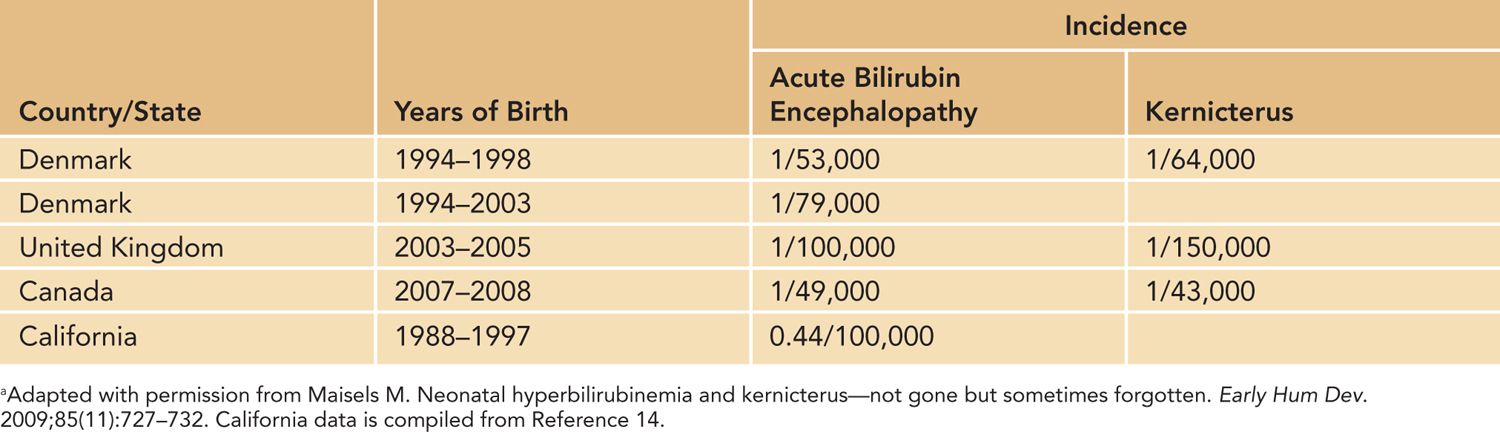
Kernicterus in Westernized Countries
Series of affected neonates from Westernized countries have recently been published from the United States,7 Canada,15 the United Kingdom and Ireland,16 and Denmark.17 The US-based Kernicterus Registry has details of 125 infants who actually developed kernicterus. In addition to cases of kernicterus, the Canadian, UK and Ireland, and Danish studies also included infants with extreme hyperbilirubinemia but without evidence of ABE. The Canadian survey further included newborns who had undergone exchange transfusion. These reports shared several common epidemiologic and etiologic features, summarized in Table 30-2. It is of note that many of the infants had been discharged as healthy from birth hospitalization but were subsequently readmitted for extreme hyperbilirubinemia. Black ethnicity and minority groups were also overrepresented, relative to the home population, in the US and UK/Ireland groups.
Table 30-2 Major Epidemiologic and Etiologic Factors for Severe Hyperbilirubinemia Common to Recent Series from Westernized Countriesa
ABO heterospecificity
G-6-PD deficiency
Other isoimmunizations
Late prematurity
Breast feeding
Sepsis
Male gender
Discharge prior to 48 hours
aFrom References 7 and 15–17.
In an Italian survey of 109 level III neonatal units, 16 cases of kernicterus (11 of these in term infants) were reported18 during the 10 years prior to 2010. A national surveillance system in Germany uncovered 11 cases of kernicterus in infants born between 2003 and 2005.19 Late prematurity and readmission of previously healthy babies were commonly encountered in this series.
Kernicterus in the Non-Western World
Kernicterus continues to occur in countries to which G-6-PD deficiency is indigenous, as well as in developing countries with underdeveloped health services, or in war zones, at a high rate. Perhaps the most devastating example emanates from Baghdad, Iraq.20 Of 162 hyperbilirubinemia-related neonatal admissions, 22% had advanced ABE, 12% died within 48 hours of admission, and 21% had posticteric sequelae. Other recent reports derive from Nigeria (115 babies with ABE, of whom 42 [36.5%] died)21; Oman (14 cases, of whom 4 [28.5%] died)22; and Turkey (10 G-6-PD-deficient neonates, of whom 5 [50%] developed kernicterus).23 In Kuwait, newborns with kernicterus were reported following traditional henna applications to their skin.24 Gamaleldin et al recently reported 249 newborns admitted during a 12-month period with a TSB level 25 to 76.4 mg/dL to Cairo University Children’s Hospital.25 Forty-four (18%) had evidence of ABE on admission; another 55 (22%) had subtle evidence of bilirubin-induced neurologic dysfunction (BIND). Twenty-six (10.4%) died, with the deaths attributable to bilirubin neurotoxicity. The most common cause for the hyperbilirubinemia was ABO incompatibility (24%), followed by Rh isoimmunization (8.8%) and G-6-PD deficiency (8.1% of 86 tested).
Surrogates for Assessing the Incidence of Kernicterus
As kernicterus is generally rare, it is difficult to assess the incidence in any specific population group with any precision. To assess the potential for developing ABE in any population groups, surrogates have been sought, including the incidence of extreme hyperbilirubinemia (serum total bilirubin [STB] > 25 mg/dL or 30 mg/dL) or the incidence of readmission for hyperbilirubinemia.14,26 The reported range for readmission27–37 for hyperbilirubinemia lies between 0.17% and 3.2%. The main reasons for readmission include lower gestational age, early discharge, unsuccessful breast feeding, and lack of predischarge assessment of the risk for subsequent hyperbilirubinemia. Punaro et al, in Brazil, recently emphasized an important effect of late prematurity on the rate of readmission for hyperbilirubinemia.38
Newman et al assessed the incidence of newborns developing extreme hyperbilirubinemia within a health care system. Despite the close surveillance and ready availability of treatment, 0.14% of newborns born at Kaiser Permanente centers in northern California developed an STB greater than 25 mg/dL; 0.01% had STB values ranging from 30.5 to 45.5 mg/dL.39 Chou et al, after controlling for gestational age, sex, maternal race, feeding type, and maternal age, compared their data from the Henry Ford Health System, Detroit, Michigan, with those of Newman et al.40 They found that the newborns under their care were less likely to have severe hyperbilirubinemia, defined as a TSB value greater than 20 mg/dL (0.6% vs 2%).40 They attributed this difference to a rigorous bilirubin screening, follow-up, and treatment program. Under less-vigorous scrutiny, the incidence of extreme hyperbilirubinemia could be expected to be even higher.
DEFINING HYPEBILIRUBINEMIA
The Hour-Specific Bilirubin Nomogram
A major advance in our understanding of bilirubin dynamics during the first week of life has been the development of the hour-specific bilirubin nomogram.41 As there are rapid changes in STB during the first postnatal days, it is inappropriate, for the purpose of bilirubin assessment, to regard units of time in days of life, but rather in hours. The higher the bilirubin value plots for postnatal hour, the higher becomes the chance of that infant developing subsequent hyperbilirubinemia. Examination of this graph will reveal an increase in STB during the first days of life, with a plateau at about 5 days. As a result, within the normal distribution of bilirubin values, no single bilirubin value can be designated to define hyperbilirubinemia. Whereas an STB value of 10 mg/dL at 12 hours of life lies above the 95th percentile, in the high-risk zone and predictive of severe hyperbilirubinemia, the same value at age 84 or 96 hours will be of little consequence. It is clear that all bilirubin values should be plotted on the nomogram to assess these values in relation to the baby’s age.
Definition of Hyperbilirubinemia
The 95th percentile on the bilirubin nomogram has been used by some in recent years to define hyperbilirubinemia,11 and several clinical research studies have incorporated this definition.42–44 This definition may not be universally practical, as the American Academy of Pediatrics (AAP, 2004) guidelines indicate the need for phototherapy below that level in cases of premature neonates or those with risk factors.45 Use of the 75th percentile for hour of life, to designate clinically significant jaundice,46 or STB values approaching the AAP indications for phototherapy47 may be viable alternatives.
PATHOPHYSIOLOGY
Bilirubin Formation and Metabolism
Bilirubin is formed from the metabolism of heme (Figure 30-1). The majority of the heme derives from the continual turnover of red blood cells (RBCs), releasing hemoglobin (Hb), although some other sources, such as myoglobin, also contribute to the heme pool. Heme is catabolized by heme oxygenase to biliverdin, by which process equimolar quantities of CO are released. Biliverdin is then reduced to bilirubin via bilirubin reductase. This form of bilirubin, also known as unconjugated or indirect bilirubin, complexes reversibly with serum albumin and in this form is transported to the liver. Intrahepatocytic uridine diphosphate (UDP)–glucuronosyltranferase 1A1 controls the conjugation of bilirubin to glucuronic acid, to form the mono- and diglucuronide forms of conjugated bilirubin. This water-soluble, polarized bilirubin form is now transported into the bile by the canalicular adenosine triphosphate (ATP)–dependent transport protein MRP249 and thence to the intestine from which most is excreted. Some bilirubin, however, may be reconverted to the unconjugated form by intestinal bacteria, thereby allowing its reabsorption and return to the bilirubin pool (enterohepatic circulation). This process is facilitated by the presence of the enteric mucosal enzyme β-glucuronidase in the newborn.
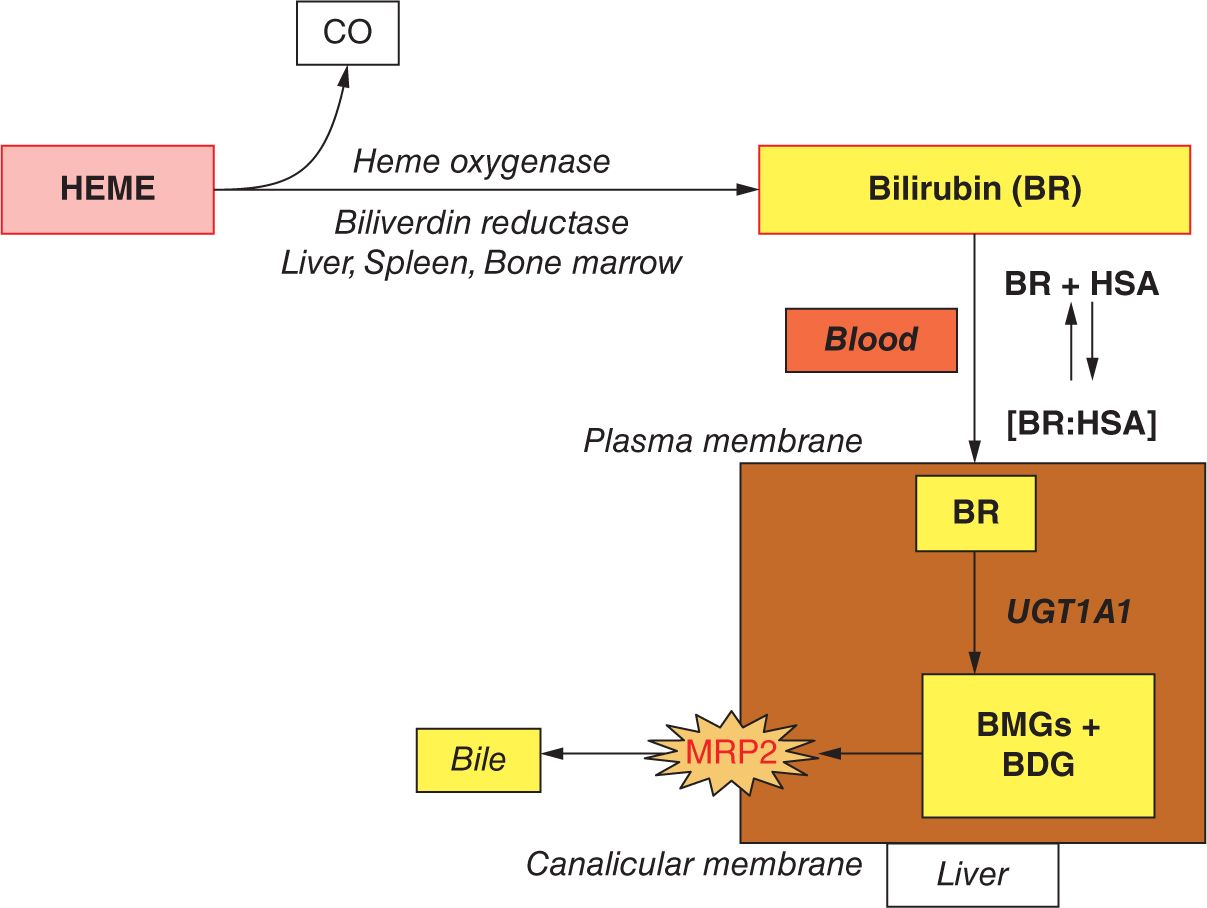
FIGURE 30-1 Main steps in the formation and elimination of bilirubin. BMG and BDG, bilirubin monoglucuronide and diglucuronide, respectively; BR, unconjugated bilirubin; HSA, human serum albumin; UGT1A1, uridine disphosphoglucuronosyl transferase 1A1. (Reproduced with permission from McDonagh AF. Controversies in bilirubin biochemistry and their clinical relevance. Semin Fetal Neonatal Med. 2010;15(3):141–147.)
Several of the steps noted have important clinical implications.
Release of CO
Carbon monoxide released concomitant with heme breakdown combines with Hb to form carboxyhemoglobin (COHb). As the CO is released in equimolar quantities with the biliverdin, quantification of COHb or end-tidal CO, corrected for ambient CO, represents the endogenous production of CO, which can be used as an index of the rate of heme catabolism, or hemolysis.50 Unfortunately, there is no bedside tool currently available for clinical use.
Heme Oxygenase
Heme oxygenase 1 (HO-1) controls the first step in heme catabolism and is the rate-limiting factor in the conversion of heme to bilirubin. Polymorphisms of the HO-1 gene encoding HO-1 may have an effect on the bilirubin production rate. Although variations in the gene promoter have been associated with higher TSB levels in adults,51,52 no effect of HO-1 polymorphisms has been reported in association with neonatal hyperbilirubinemia.53 The enzyme may be important with regard to bilirubin pharmacotherapeutics. Synthetic heme analogues, metalloporphyrins, are competitive inhibitors of HO-1 and have been used clinically to inhibit the enzyme activity, thereby either preventing bilirubin formation or limiting further bilirubin production when treating established hyperbilirubinemia.54 Some studies have documented successful use of metalloporphyrins, as reviewed,54 but the Food and Drug Administration (FDA) has not yet approved the drug for routine clinical use.
Bilirubin Binding and Unbound Bilirubin
As long as the unconjugated bilirubin is bound to serum albumin, this complex is thought to be unable to cross the blood-brain barrier and enter the brain cells. Unbound bilirubin, on the other hand, is potentially neurotoxic and has the ability to cause neuronal damage. Indeed, the unbound bilirubin fraction concentration is a much better predictor of bilirubin neurotoxicity than the STB.55,56 Unfortunately, there is currently no bedside tool to make unbound bilirubin measurement readily available or clinically useful, and the bilirubin-albumin ratio is sometimes used as a surrogate to determine the need for exchange transfusion.45 Some factors that may facilitate the finding of increased serum unbound bilirubin and therefore precipitate the penetration of bilirubin into brain cells include hypoalbuminemia, asphyxia, hypothermia, metabolic acidosis, and sepsis.
Uptake Genes
Uptake of bilirubin into the liver is controlled by the solute carrier organic anion transporter protein 1B1, SLCO1B1, also known as OATP2.57 This sinusoidal transporter facilitates the hepatic uptake of bilirubin as well as other substances, and varying expression of the gene may affect bilirubin kinetics and metabolism. SLCO1B1∗1b variant is associated with neonatal hyperbilirubinemia in Tawainese newborns, especially when coupled with UGT1A1 (uridine disphosphoglucuronosyl transferase 1A1) variants.58 In a US-based study, coexpression of SLCO1B1∗1b with G-6-PD A-was associated with hyperbilirubinemia.59 These studies and others60 emphasized the importance of gene interactions and ethnic variation in the pathogenesis of hyperbilirubinemia.
Genetic Control of UGT1A1
The function of the UGT enzyme is to conjugate glucuronic acid to certain target substrates to facilitate their elimination from the body. Of major importance to the conjugation and elimination of bilirubin is the UGT1A1 gene,61 situated on chromosome 2q37. This gene encodes the UGT1A1 enzyme and is therefore of paramount importance in bilirubin conjugation. The gene consists of 4 common exons (exons 2, 3, 4, and 5) and 13 variable exons, but of the latter, only variable exon A1 is of importance regarding bilirubin conjugation (Figure 30-2). The variable exon A1 functions in conjunction with the common exons to regulate the synthesis of the individual enzyme isoform. Upstream of each variable exon is a regulatory noncoding promoter that contains a TATAA box sequence of nucleic acids. Mutations or polymorphisms of the variable exon A1, its promoter, or the common exons 2–5, may interfere in the normal process of bilirubin conjugation. While polymorphisms of the noncoding promoter area may affect bilirubin conjugation by diminishing expression of a normally structured enzyme, mutations of the coding areas may alter the structure of the enzyme molecule, thereby interfering with or abolishing enzyme function.
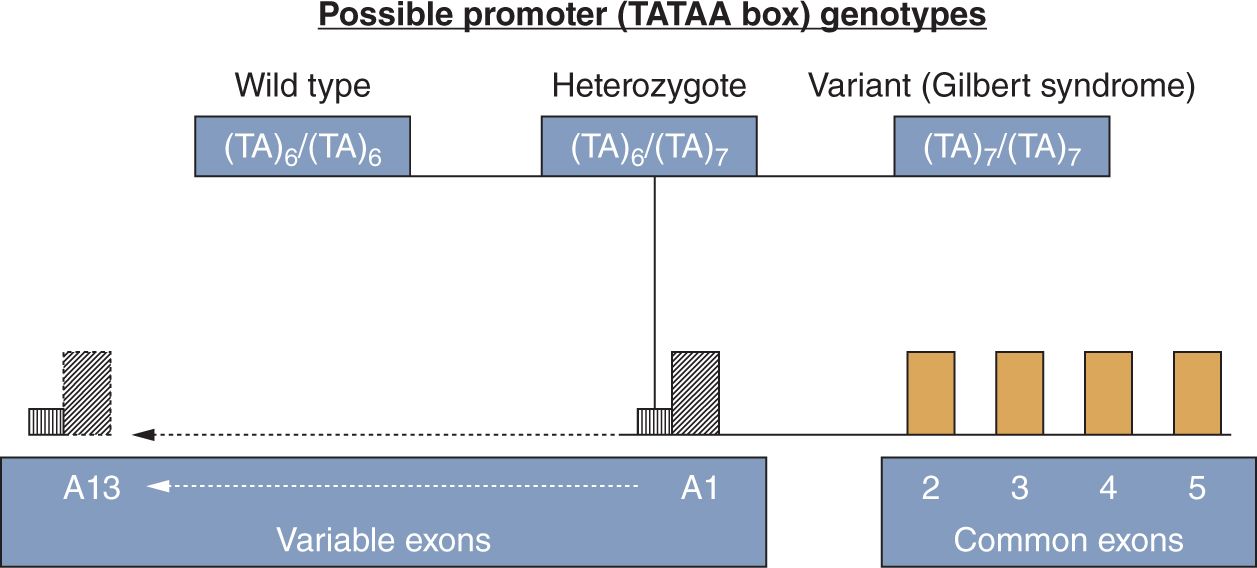
FIGURE 30-2 The uridine diphosphate (UDP)–glucuronosyltransferase gene. For explanation, see the text on the genetic control of UGT1A1. (From Ref. 62.)
Imbalance Between Bilirubin Production and Elimination
The STB concentration at any point reflects 2 major processes: bilirubin production and bilirubin elimination.63 Most important is the concept of lack of equilibrium between bilirubin production and conjugation. As long as these processes remain in equilibrium, the STB should remain within normal limits. In the postnatal period, increased heme catabolism in combination with diminished UGT1A1 activity results in imbalance between these processes with increasing bilirubin levels. The enterohepatic circulation may add to the bilirubin load. Should this imbalance remain mild or moderate, STB concentrations should not exceed the 95th percentile on the hour-of-life-specific bilirubin nomogram.41 However, should bilirubin production exceed its elimination, hyperbilirubinemia may occur. Some degree of increased heme catabolism, not necessarily severe, occurs in many cases of hyperbilirubinemia, even when no obvious hemolytic etiology is apparent.42,46,64 Despite increased hemolysis and production of large amounts of bilirubin, some babies may not develop increased STB because their hepatic bilirubin-conjugating capacity is mature. On the other hand, minimally increased hemolysis in the face of immature bilirubin conjugation may well result in hyperbilirubinemia. This concept has been likened to a sink of water. Increased inflow relative to the drainage will result in a rise in the water level, whereas increased inflow along with an efficient drainage system will result in minimal rise of the water level.63 Kaplan et al demonstrated this concept mathematically.65 Using blood carboxyhemoglobin corrected for ambient CO (COHbc) to index heme catabolism as the numerator of the equation and serum-conjugated bilirubin to reflect bilirubin conjugation as the denominator, they found that the combined effect of bilirubin production and conjugation correlated better with TSB levels than any of these processes individually.
Increased Danger Associated with Hemolysis
It is generally believed that neonates with hemolytic disease are at a higher risk of bilirubin neurotoxicity than those whose hyperbilirubinemia is not the result of hemolysis.66,67 Indeed, many original reports of kernicterus emanated from infants with Rh isoimmunization.68 Data from a few studies are supportive of this concept. In a Turkish study, a positive DAT (or direct Coombs test), due to Rh isoimmunization or ABO incompatibility, was used as a presumed marker of hemolysis.69 In children with indirect hyperbilirubinemia, DAT positivity was associated with lower IQ scores and a higher incidence of neurologic abnormalities than controls without a positive test but with similar bilirubin concentrations. In DAT-positive Norwegian males who had TSB levels greater than 15 mg/dL for longer than 5 days, IQ scores were significantly lower than average for that population.70 In the Jaundice and Infant Feeding Study, 5-year IQ values of infants with TSB greater than 25 mg/dL in combination with a positive DAT were significantly lower than hyperbilirubinemic counterparts but with a negative DAT (−17.8 IQ points, 95% confidence interval [CI] −26.8 to −8.8).71 In a reanalysis of the data from the Collaborative Perinatal Project, Kuzniewicz and Newman found the presence of a positive DAT in those with a TSB of greater than 25 mg/dL was associated with a 6.7-point decrease in IQ scores.72 An increase in the duration of exposure to high TSB levels was also associated with an increase in neurologic abnormalities. Hemolytic conditions were among the most common etiologies for hyperbilirubinemia in the previously mentioned neonates with ABE recently reported from Egypt: ABO incompatibility (24%), Rh isoimmunization (8.8%), and G-6-PD deficiency (8.1% of 86 who were tested).25
The reason for hemolysis increasing the risk for bilirubin neurotoxicity is not clear. Rapid rise in STB and earlier onset of the peak bilirubin value may contribute to this effect. Rapid saturation of the extravascular tissues with bilirubin may further increase the intravascular component. The AAP recommends institution of treatment of hyperbilirubinemia at lower levels of STB in newborns in whom hemolysis is recognized than in nonhemolyzing controls.45
THE DREADED COMPLICATION OF EXTREME HYPERBILIRUBINEMIA: ACUTE BILIRUBIN ENCEPHALOPATHY OR KERNICTERUS
Definitions
The term kernicterus is sometimes used interchangeably for acute encephalopathy and the chronic form of choreoathetotic cerebral palsy. The clinical picture of these conditions, however, is very different.73 Bilirubin encephalopathy implies the central nervous system findings that result from bilirubin toxicity to the basal ganglia and other brainstem nuclei.45 The Subcommittee on Hyperbilirubinemia of the AAP recommends using the term acute bilirubin encephalopathy to denote the acute manifestations of bilirubin toxicity seen in the first few weeks after the hyperbilirubinemic episode. The term kernicterus, in contrast, should be used in association with the chronic and permanent clinical sequelae of bilirubin toxicity.45 The expression bilirubin-induced neurologic dysfunction (BIND) usually refers to a form of bilirubin neurotoxicity that is subtle, including neurodevelopmental and auditory disabilities that appear to be due to bilirubin toxicity but without the classic findings of kernicterus.74,75
Pathology of Bilirubin Toxicity
The pathologic hallmark of bilirubin neurologic damage is staining and necrosis of neurons in the basal ganglia, hippocampal cortex, subthalamic nuclei, and cerebellum followed by gliosis of these areas in survivors (Figure 30-3). The cerebral cortex is generally spared.
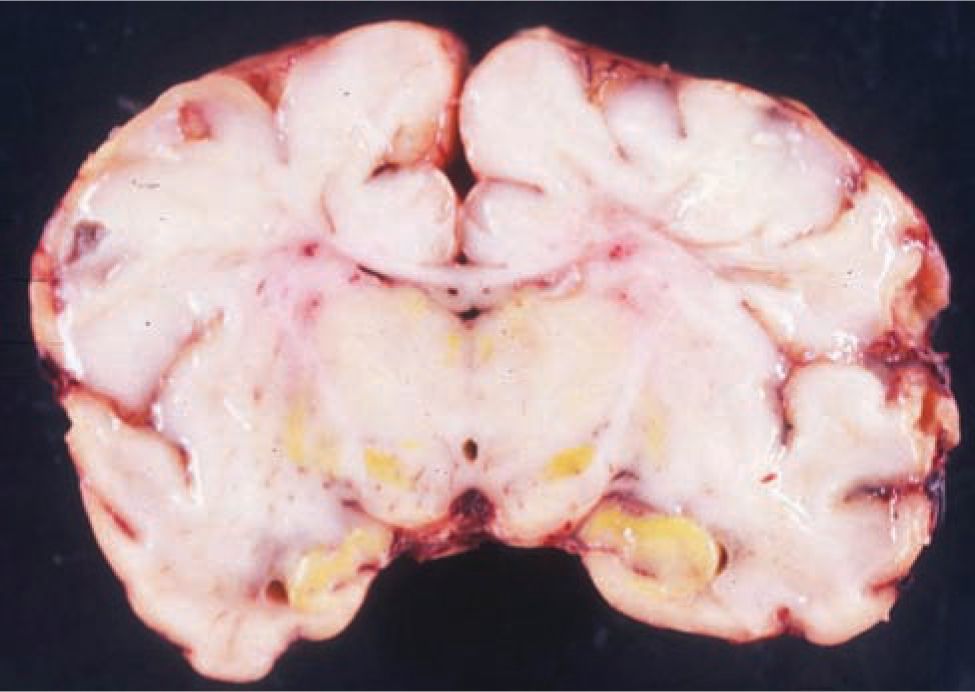
FIGURE 30-3 Bilirubin (yellow) staining of the basal ganglia of a newborn who died from acute bilirubin encephalopathy. (Reproduced with permission of Zangen S, Kidron D, Gelbart T, et al. Fatal kernicterus in a girl deficient in glucose-6-phosphate dehydrogenase: a paradigm of synergistic heterozygosity. J Pediatr. 2009;154(4): 616–619.)
Clinical Picture of Kernicterus
Acute Bilirubin Encephalopathy
Early clinical features of ABE include with severe lethargy and poor feeding (phase 1). Although these signs are nonspecific, in the presence of severe hyperbilirubinemia, encephalopathy should be suspected to avoid delay in institution of therapy. During the middle of the first postnatal week (phase 2), muscle tone may fluctuate between hypo- and hypertonia, and a high-pitched cry develops. Spasm of the extensor muscles with back arching, opisthotonus, retrocollis, and impairment of upward gaze resulting in the “setting sun sign” ensue in phase 3, with fever, seizures, apnea, and death following45,73,77 (Figure 30-4).
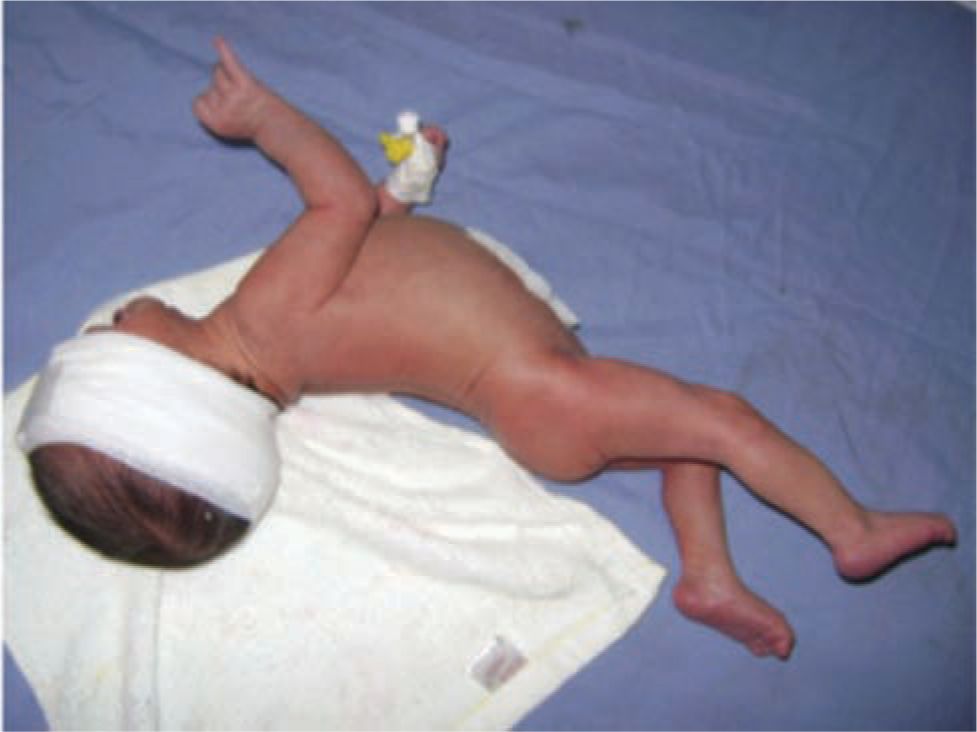
FIGURE 30-4 Baby with acute bilirubin encephalopathy. Note arching of extensor muscles of back and neck. (Reproduced with permission from Bhutani VK, Stevenson DK. The need for technologies to prevent bilirubin-induced neurologic dysfunction syndrome. Semin Perinatol. 2011;35(3):97–100.)
Chronic Kernicterus
The clinical picture of kernicterus in its chronic form has been well described.73 Affected individuals may display a dystonic or athetoid movement disorder, an auditory processing disturbance that may be associated with hearing loss, motor ocular impairment of upward gaze, enamel dysplasia of the teeth, and hypotonia and ataxia due to cerebellar involvement. Many are unable to walk, require a feeding tube, or have other feeding disabilities. Mental retardation is not uncommon, but some do not have evidence of mental impairment. Hearing and visual impairments occur frequently. Motor spasticity, ataxia, and dyskinesia or hypotonia may be seen.14,45
Subtle Bilirubin Encephalopathy and Auditory Neuropathy
Bilirubin encephalopathy may not always manifest as the classic, chronic picture of kernicterus. In some, bilirubin-induced neurological damage may result in subtle bilirubin encephalopathy, or BIND. These children have less-severe injury than those with classic kernicterus but may have cognitive disturbances, mild neurologic abnormalities, isolated hearing loss, or auditory neuropathy.74,79 Auditory neuropathy associated with hyperbilirubinemia is not simply a sensorineural hearing loss, but rather is the result of dysfunction at the level of the auditory brainstem or nerve. Functionally, auditory neuropathy or dyssynchrony is characterized by absent or abnormal brainstem auditory evoked potentials, but with normal inner ear function. In these cases, hearing screening utilizing automated auditory brainstem responses will identify the condition. Evoked otoacoustic emission studies, reflecting inner ear function, may be normal, however, and if used alone, may result in the auditory neuropathy being missed. Awareness of bilirubin auditory neuropathy is of practical importance as cochlear implantation has been used successfully in children with this condition.74 The mechanism by which hearing is improved is not clear, as the implant is actually proximal to the neural lesion.
DIFFERENTIAL DIAGNOSIS
A list of conditions associated with neonatal jaundice can be seen in Table 30-3. Some specific conditions, chosen because they are frequent, demonstrate a principle, or have public health implications, have been selected for further description in the following paragraphs.
Table 30-3 Common Conditions Associated with Neonatal Jaundice
General
Physiological jaundice
Breast milk jaundice
Hemolytic conditions
Immune
Rh
ABO
Other immunizations
Nonimmune
G-6-PD deficiency
Hereditary spherocytosis
Pyruvate kinase deficiency
Red cell membrane defects
Unstable hemoglobinopathies
Conditions associated with diminished bilirubin conjugation or elimination
Gilbert syndrome
Crigler-Najjar syndromes types 1 and 2
Hypothyroidism
Breast milk jaundice
Prematurity or late prematurity
Physiological Jaundice
About 60% of term newborns and even more breast-feeding infants will develop some degree of visible jaundice. During the first postnatal week, there is a natural increase in STB, reaching a mean peak of 5 to 6 mg/dL at about 5 days. Visible jaundice occurring during the first 24 hours should never be regarded as physiologic.45 Any bilirubin concentration greater than the 95th percentile should not be regarded as physiologic. Following the fifth day, the STB values decrease gradually.
Physiologic jaundice occurs because of increased heme catabolism in combination with diminished bilirubin conjugation, even in the absence of known risk factors for jaundice. Newborn babies have a high rate of heme catabolism, the result of shorter RBC lifespan and greater hematocrit and RBC volume than older counterparts. Also, UGT1A1 enzyme activity is immature in newborns, leading to bilirubin buildup in the immediate postnatal period.80 This physiologic phenomenon can be exacerbated by breast feeding (below) or increased reabsorption of bilirubin from the bowel in newborns by way of the enterohepatic system, adding to the already-overloaded bilirubin pool and putting further strain on the limited conjugative system.
Jaundice in Breast-Feeding Infants
Healthy term and late-preterm infants who are breast-feeding have higher STB concentrations than formula-feeding counterparts. Some authorities recognize 2 specific entities with regard to jaundice in breast-fed infants, the first, related to inadequate feeding, leading to mild dehydration (breast feeding jaundice), and the second due to breast milk itself (breast milk jaundice).81 Breast-feeding jaundice occurs during the first postnatal week. Many of these babies have excessive weight loss, the result of delayed initiation of breast feeding, insufficient frequency of feeding, and supplementation with water. These factors may result in increased bilirubin absorption via the enterohepatic circulation. Frequent and successful breast feeding not only will enhance the milk supply, thereby increasing caloric intake and providing sufficient fluid, but also will accelerate intestinal transit time and facilitate excretion of meconium, thereby reducing the enterohepatic bilirubin circulation. Phototherapy may be necessary. In contrast, breast milk jaundice appears after the first week. The infants are healthy and vigorous and have been gaining weight appropriately. It is postulated that substances present in the breast milk may inhibit UGT1A1 activity. Suggested substances include the progesterone metabolite pregnane-3-alpha-20-beta-diol or beta-glucuronidase. It has been demonstrated that presence of the (TA)7 UGT1A1 promoter polymorphism (UGT1A1∗28) may be responsible for prolonged jaundice in breast-feeding newborns.82 Most infants with breast milk jaundice will respond to phototherapy, and in most cases, it is unnecessary to discontinue breast feeding during treatment.
Hemolytic Conditions
Neonatal hemolytic conditions may be divided into immune and nonimmune pathophysiologic subgroups (Table 30-4). Although Rh isoimmunization is today largely preventable, much of our knowledge regarding the pathophysiology of kernicterus derives from the study of babies affected with this hemolytic condition. ABO immune disease is now the most common immune condition encountered. Of the nonimmune causes, G-6-PD deficiency is the most important from a public health standpoint and is associated with the development of extreme hyperbilirubinemia and, in some cases, bilirubin encephalopathy.
Table 30-4 Some Important and Commonly Encountered Risk Factors for Neonatal Hyperbilirubinemiaa
Late prematurity
Breast feeding
Early discharge
Poorly established nursing, excessive weight loss
Hemolytic conditions
Immune hemolytic disease (direct antiglobulin titer [DAT] positive)
G-6-PD deficiency
Previous sibling with hyperbilirubinemia
Bruising, cephalhematoma
Infection
aA complete list of risk factors can be found in Reference 45.
Immune Hemolytic Conditions
The Direct Antiglobulin Test
The DAT, otherwise known as the direct Coombs test, is the hallmark of isoimmunization83 and is indicative of maternally derived immunoglobulin (Ig) G directed against fetal/neonatal RBC antigenic sites. The antiglobulin reaches the fetus via the placenta. The DAT detects antibody but does not identify the specific antibody type; for this purpose, further tests are necessary. Agglutinated clumps of RBCs may be identified visually or microscopically. Although a positive DAT is potentially associated with increased hemolysis and hyperbilirubinemia, this may not be universally so. Herschel et al found that a newborn with a positive DAT had only a 59% chance of having a 12-hour corrected end-tidal CO (ETCOc) value greater than the 95th percentile, and only 14.8% of DAT-positive neonates developed a STB equal to or greater than the 75th percentile.84 Ozolek et al found a similar relationship between DAT and hyperbilirubinemia/jaundice in ABO-heterospecific mother-infant pairs.85
Rh Isoimmune Hemolytic Disease
Rh disease in pregnancy may lead to intrauterine hemolysis, severe intrauterine anemia, hydrops fetalis, and intrauterine death. Following delivery, ongoing hemolysis may result in severe hemolytic disease of the newborn (HDN), anemia, and hyperbilirubinemia with the potential for bilirubin encephalopathy.
Background: The Immunization Process
Although the Rh group comprises the C, c, D, E, and e antigens, each of which may result in isoimmunization and hemolysis, RhD isommunization was formerly the most common, and when it does occur, is still the most severe. In white populations, 13% to 15% of individuals are Rh negative, but only about half that number are encountered in African Americans, and few Asian individuals are Rh negative. Because of paternal heterozygosity (D/d), an Rh-negative mother may have an Rh-negative fetus in about 50% of pregnancies. However, because of factors such as nonuniversal fetomaternal transmission of fetal blood and variable maternal immune responses, the overall incidence of Rh isoimmunization is infrequent and reported to be 6.8 cases per 1000 live births.86
Exposure of a D-negative woman to the D antigen, occurring either antepartum or intrapartum by fetomaternal passage of fetal RBCs containing the D antigen, sets the immunization process into action. Although first-pregnancy isoimmunization has been documented, in primigravidas the process usually begins too late to allow for sufficient maternal IgG antibody to be produced and transferred across the placenta.87,88 Transfusion of Rh-positive RBCs may also occur during abortion, blood administration, amniocentesis, chorionic villus sampling, or fetal blood sampling, thereby sensitizing the mother. In response to the antigen, the mother’s immune system produces anti-D IgG antibodies, which cross the placenta and adhere to the D-antigen sites of the fetal RBCs. The resulting antigen-antibody response culminates in hemolysis and anemia. During subsequent pregnancies, this response may become progressively more severe and rapid. The bone marrow now releases increased numbers of circulating immature RBCs (erythroblastosis), and hepatomegaly and splenomegaly may ensue due to extramedullary hematopoiesis. Fetal hydrops, including generalized tissue edema and pleural, pericardial, and peritoneal effusions, may result. Formation of this extravascular fluid results from hypoproteinemia, tissue hypoxia, and capillary leakage, along with congestive cardiac failure, itself the result of poor myocardial function and diminished cardiac output due to anemia and venous congestion.89
Management of the Pregnancy
Hydrops fetalis is associated with a high mortality rate. Should the fetus become anemic, the option to perform intrauterine transfusion must be weighed against delivery. This decision will depend primarily on the gestational age: With increasing maturity, the potential for inherent complications involved in preterm delivery will decrease relative to the dangers involved in performing intrauterine transfusion. Aminocentesis-based regimens for detecting fetal anemia90 are being replaced by a combination of advancing ultrasonographic techniques and developing genetic technologies, as described further in this chapter. Furthermore, modern advances in DNA technologies allow for accurate determination of paternal RhD gene status; cell-free fetal DNA determination techniques may allow for minimally invasive determination of fetal Rh type in a maternal blood sample.91–93 Administration of rhesus immune globulin during pregnancy to all nonimmunized, RhD-negative women, in combination with routine postpartum administration of the globulin to those delivering an Rh-positive newborn,94 has reduced the incidence of antenatal immunization from 14% to 0.1%. The globulin should also be administered following spontaneous or elective abortion, amniocentesis, chorionic villus sampling, or fetal blood sampling. In developing countries where Rh immune prophylaxis is not yet routine, the incidence of Rh HDN in still common.95
Determination of the maternal anti-D titer to find the critical titer associated with a high risk of kernicterus is an important step in the monitoring of an RhD-sensitized woman. The critical titer89 ranges from 1:8 to 1:32. Doppler assessment of the blood flow velocity in the middle cerebral artery of the fetus is replacing amniocentesis in the detection of fetal anemia, the fetal anemia resulting in increased blood flow velocity due to decreased blood viscosity and increased cardiac output.96 Should the results suggest anemia, fetal blood is then sampled by cordocentesis. Intrauterine transfusion is an option when the hematocrit is less than 30% and the fetus less than 35 weeks’ gestation. However, if the pregnancy has reached 35 weeks’ gestation or more, delivery will be prudent. In experienced hands, the outcome of intrauterine transfusion should be good.97
Postnatal Management of the Newborn
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


