Cystic Lung Lesions
INTRODUCTION
Congenital cystic lung disease covers a spectrum of lung malformations that include congenital cystic adenomatoid malformation (CCAM; which is also defined by the term congenital pulmonary airway malformation or CPAM), bronchopulmonary sequestration (BPS), congenital lobar emphysema (CLE), and bronchogenic cysts. The intricate process of lung development begins at about 4 weeks’ gestation and consists of 6 discrete phases: (1) the embryonic stage (4–7 weeks’ gestation); (2) the pseudoglandular stage (5–17 weeks’ gestation); (3) the canalicular stage (16- to 26-weeks’ gestation); (4) the saccular stage (24- to 38-weeks’ gestation); (5) the alveolar stage (36-weeks’ gestation to 2 years of age); and (6) microvascular maturation (birth to 2–3 years of age).1
A key stage in the development of congenital cystic lung disease appears to be the pseudoglandular stage, when branching morphogenesis occurs. Branching morphogenesis results in the formation of the conductive airway system, which is an essential prerequisite to alveolization. Lung development is influenced at all stages by the spatial and temporal distribution of a variety of signaling molecules and their receptors and by the positive and negative control of signaling by paracrine, autocrine, and endocrine mechanisms. It is likely that a developmental arrest or maturation disorder in the pseudoglandular stage is responsible for the vast majority of developmental lung anomalies, and although many of the responsible genes and gene products have been identified, the precise mechanisms involved are unknown.2–5 Analysis of resected fetal lung lesions have shown an imbalance between cellular proliferation and apoptosis, suggesting a loss of growth regulation.6
Although there is a tendency to compartmentalize congenital cystic lung disease into discrete pathological conditions, there is considerable overlap between conditions. An example is the shared features of CCAM and BPS within so-called hybrid lesions, which exhibit the histological features of CCAM combined with the anomalous vascular anatomy of BPS.
Epidemiology of Neonatal Cystic Lung Lesions
There are few data on the epidemiology of cystic lung lesions. One frequently quoted study using population-based data estimated the incidence of CCAM to be 1 per 25,000 to 35,000 pregnancies.7 Most cystic lung lesions occur with equal frequency in the right and left lungs and have a slight preponderance for males.8
Differential Diagnosis
The differential diagnosis of cystic lung lesions in an infant include CPAM, BPS, CLE, bronchogenic cysts, as well as congenital diaphragmatic hernia (CDH) and a variety of congenital and acquired cystic lesions of the lung and mediastinum. Some of the acquired cystic lung conditions include pulmonary interstitial emphysema, postinfectious pulmonary cysts, lung abscesses, and rare tumors, including bronchioloalveolar carcinoma (BAC) and pleuropulmonary blastoma (PPB).
CONGENITAL CYSTIC ADENOMATOID MALFORMATION AND CONGENITAL PULMONARY AIRWAY MALFORMATION
The CCAM and CPAM cystic lung hamartomas are associated with a proliferative increase in terminal bronchioles relative to alveoli. The result is an intrapulmonary mass that is usually confined to 1 lobe of the lung. The nomenclature and descriptive classification of these lesions has changed significantly since the original description in 1949 of a “congenital adenomatoid lesion” of the lung.9 In 1977, Stocker et al proposed a classification scheme (types I, II, and III) based on the clinicopathologic attributes of CCAMs.10 The most common type (type I) accounts for over 50% of cases and is characterized by single or multiple cysts larger than 2 cm. Based on the increased frequency of prenatal diagnosis and the realization that the natural history of a fetal lung lesion could be predicted by its sonographic appearance, Adzick et al proposed a classification system that defined CCAMs as either microcystic (appearing solid or “echogenic” on ultrasound and more often associated with fetal hydrops) or macrocystic, with definitive cysts larger than 5 mm in diameter.11 More recently, the term congenital pulmonary airway malformation has been coined that provides a unifying classification of lung malformations into CPAM 0, 1, 2, 3, and 4 based on the site of suspected malformation development along the tracheobronchial/acinar structural unit pathway.12 In this classification system, the vast majority of malformations are CPAM 1 (Figure 28-1), implying that these are developmentally derived from the bronchial or bronchiolar units. The relationship between these 2 nomenclature systems is summarized in Table 28-1.
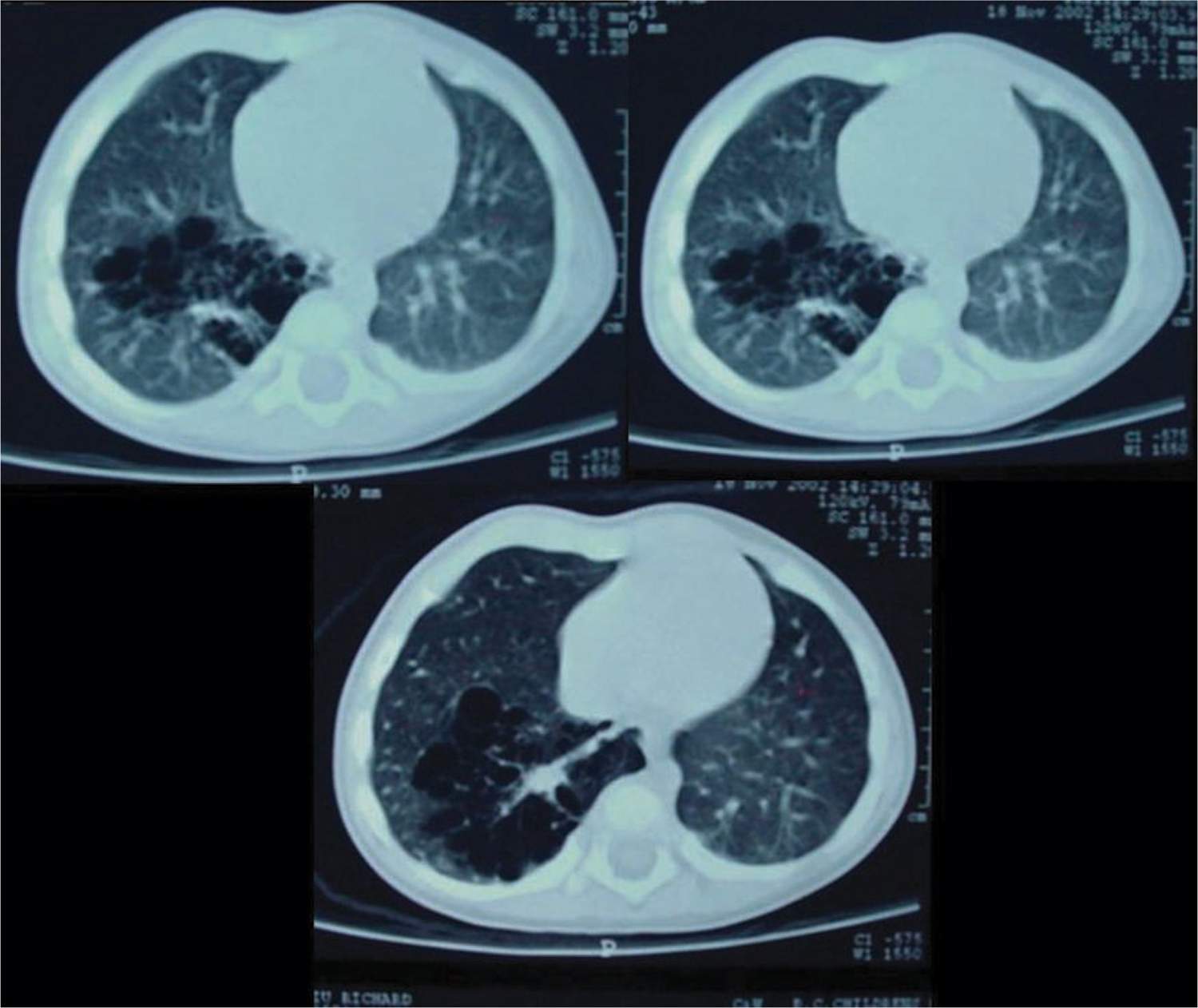
FIGURE 28-1 Computed tomographic scan showing macrocystic, multilocular congenital pulmonary airway malformation type 1.
Table 28-1 Comparisons of Features of CPAM and CCAM Subtypesa
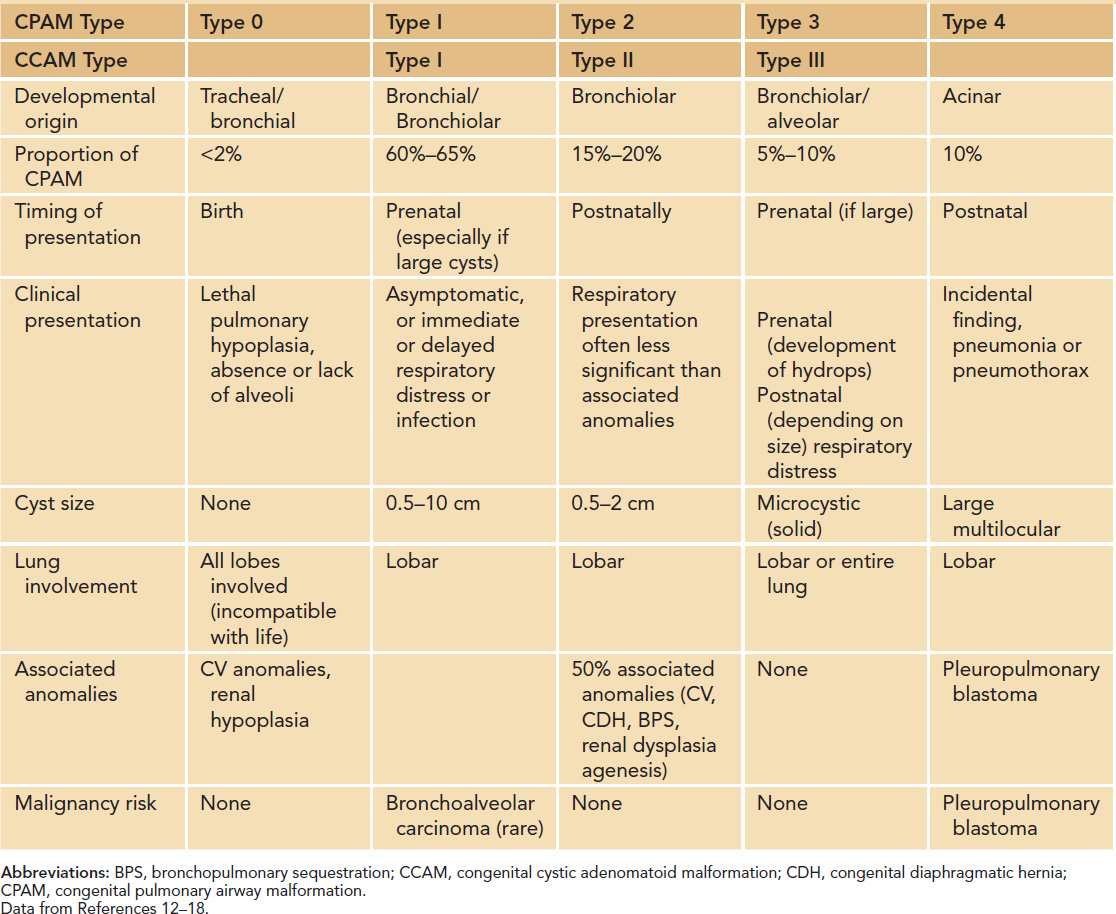
CPAM 1 accounts for 60% to 65% of postnatal cases.12 It arises from a single lobe (Figure 28-2) and consists of single or multiple large, intercommunicating cysts that are surrounded by smaller cysts and atelectatic normal lung. The cyst walls are thin and lined by an epithelium that varies from low cuboidal to ciliated pseudostratified columnar epithelium. The walls contain fibromuscular connective tissue that in some areas forms a muscular wall resembling bronchi. Cartilage islands may be present within the walls along with arteries and veins, which are classically derived from the pulmonary circulation. There are rare case reports of adenocarcinoma developing in association with a CPAM 1, suggesting the possibility of a preinvasive lesion that becomes cancerous over time.13
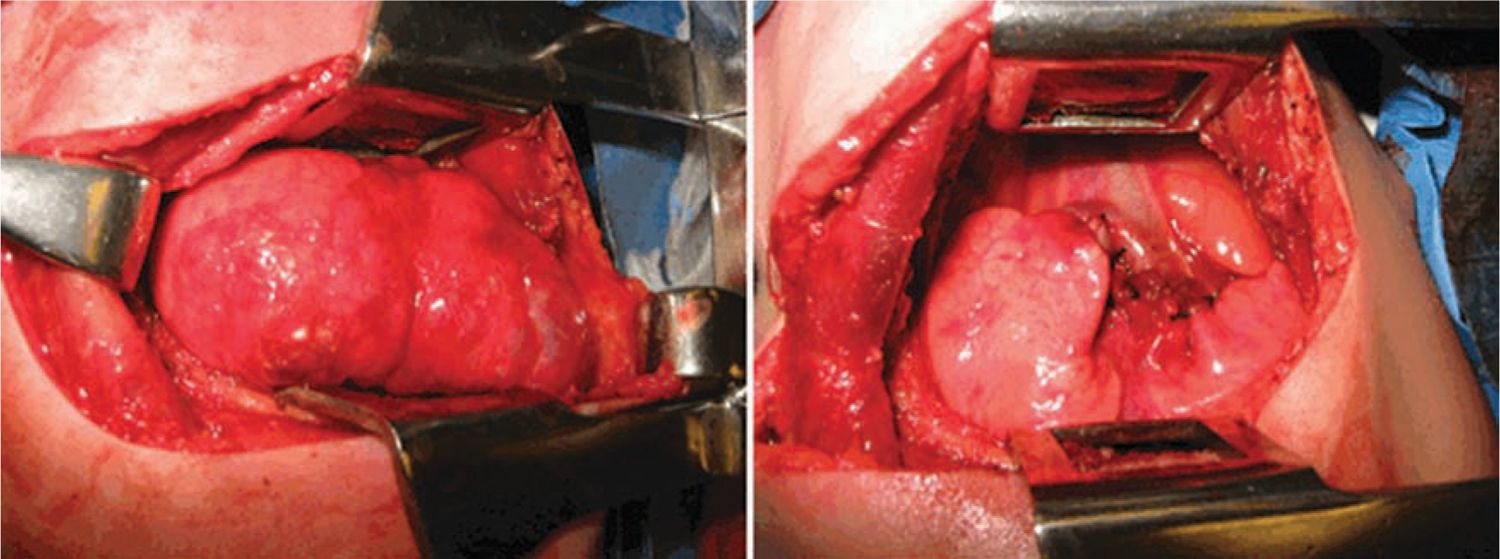
FIGURE 28-2 Operative photograph demonstrating large type congenital pulmonary airway malformation arising from right upper lobe, pre- (left panel) and postresection (right panel).
CPAM 2 lesions, which account for about 20% of postnatally diagnosed cases, contain numerous smaller cysts (less than 2 cm), which appear as “back-to-back” bronchiole-like structures lined by a simple cuboidal to columnar epithelium. A key feature of CPAM 2 lesions is their association in 50% of cases with other malformations, including CDH, cardiovascular abnormalities, and rarely lethal malformations such as bilateral renal agenesis or dysplasia.14,15
CPAM 3 represents the microcystic variant, which is now recognized antenatally as being at greatest risk for the development of hydrops.16 The microscopic appearance is that of irregular bronchiole-like structures lined by cuboidal epithelium.
CPAM 4 accounts for about 10% of cases and results when disordered acinar development produces a hamartomatous cyst of variable size in the lung periphery. The cysts are lined by types 1 and 2 alveolar cells.12 The clinical presentation may be variable (asymptomatic, pneumonia, spontaneous pneumothorax).
An area of controversy has been an understanding of the relationship between CPAM and PPB. The clinical, radiologic, and even histologic overlap between cystic PPB and CPAM 4 is well recognized, and for a time it was thought that PPB resulted from “malignant transformation” of a CPAM. However, that view has been discounted by thorough histological characterization and subtyping of PPB,17 and it is now accepted that CPAM and PPB represent pathologically discrete entities.18
Fetal Diagnosis and Outcome
With advances in ultrasound technology and widespread screening, the detection of fetal lung lesions has become increasingly common. The combination of high-resolution ultrasound, vascular Doppler, and magnetic resonance imaging (MRI) usually results in an accurate fetal lung anomaly diagnosis. A suspected fetal lung anomaly should prompt additional diagnostic testing, including echocardiography and amniocentesis for karyotyping, to exclude significant associated cardiovascular or chromosomal malformations.
Once a fetal lung malformation has been identified, it should be closely monitored because its behavior may be difficult to predict. It has been reported that approximately 40% of CCAMs will grow rapidly between 18 and 26 weeks, leading to mediastinal compression and the development of fetal hydrops.19 Conversely, the phenomenon of spontaneous regression, regardless of cyst morphology (ie, macro vs microcystic), is well documented.7,20–24 Whether CCAMs can permanently spontaneously resolve is debatable. Most apparently vanishing fetal CCAMs are detectable on postnatal computed tomographic (CT) scans25,26; however, the phenomenon of postnatal disappearance documented by CT scan has been reported.27
Fetal hydrops occurs when the rapidly growing lung mass causes vena caval compression and cardiac failure. Polyhydramnios, pleural effusions and ascites, and subcutaneous edema are all signs of fetal hydrops. The development of hydrops portends a poor prognosis for the fetus and may be an indication for fetal intervention, including catheter drainage for macrocystic disease or fetal thoracotomy and lung resection for microcystic disease. The latter requires multidisciplinary expertise in fetomaternal care and therefore should only be undertaken in specialized fetal treatment centers. A small proportion of CCAMs associated with fetal hydrops may also manifest the maternal mirror syndrome.28 In this scenario, the mother’s condition begins to mirror that of her fetus, as she develops progressive symptoms of preeclampsia. The pathophysiology of this condition is poorly understood; however, fetectomy and removal of the placenta is lifesaving for the mother.
The ability to predict which fetuses may develop hydrops has been enabled by the development of an ultrasound prediction tool called the CCAM volume-to-head-circumference ratio (CVR).19 Using an elliptical volume approximation of the CCAM that is gestationally adjusted by dividing by the head circumference, the CVR allows estimation of the risk of hydrops development prior to 28 weeks’ gestation (after which the risk of developing hydrops is significantly reduced). If the calculated CVR is greater than 1.6 and there is no dominant cyst, the risk that a fetus will develop hydrops is approximately 80%. Such fetuses should be closely monitored (twice-weekly ultrasound) for developing signs of hydrops.
Fetal Intervention
With an evolved understanding of the natural history of fetal CCAMs, the potential lifesaving benefit of fetal intervention for the development of hydrops has been realized. The options for intervention are maternal steroid administration, and fetal thoracotomy and lung resection for microcystic CCAM and ultrasound-guided thoracoamniotic shunting for macrocystic CCAM. The largest reported experience to date with maternal steroid treatment of fetal CCAM is 13 patients, all with a CVR greater than 1.6, of whom 9 had developed hydrops. Following a single dose of steroids, the CVR decreased in 8 (62%), and hydrops resolved in 7 (78%). All fetuses survived to delivery, and 11/13 (85%) survived to hospital discharge.29
For fetuses with a large CCAM and a dominant cyst, there may be a role for fetal thoracentesis and cyst aspiration or placement of a thoracoamniotic shunt. It is important to remember that fetuses with large CCAMs are also at risk of having significant pulmonary hypoplasia, so despite an apparently favorable response to maternal steroids or catheter drainage, there is the potential for marked respiratory insufficiency at birth, which may require significant ventilatory support or even extracorporeal membrane oxygenation (ECMO). In rare instances if the risk of severe pulmonary hypoplasia at birth is high, there may be a role for an ex utero intrapartum treatment (EXIT) to lung resection approach. In this highly specialized procedure, the baby is delivered by cesarean section and maintained on the placental circulation while thoracotomy and lung resection are undertaken. This approach has been used successfully in 9 high-risk cases of fetal CCAM. In this series, 8 of 9 patients survived, although 4 did require ECMO following lung resection, attesting to the severity of associated pulmonary hypoplasia.30
Postnatal Diagnosis and Management
If a prenatal diagnosis of a CCAM has been made, a decision needs to be made regarding location of delivery. Because of the unpredictability of respiratory distress at birth due to underlying pulmonary hypoplasia or worsening mediastinal shift and adjacent lung collapse caused by air or fluid trapping, any fetus with a history of a large CCAM should be delivered in a center with access to urgent pediatric surgical care. Fetuses with “disappearing” or small, peripheral cystic lesions can be safely delivered outside a specialty perinatal center as long as the baby can be expeditiously transferred to a specialty center if needed.
The assessment of a baby with a history of fetal CCAM or a baby without a prenatal diagnosis who has respiratory symptoms begins with a chest radiograph. The differential diagnosis of a postnatally diagnosed CCAM includes CDH, especially when the left side is involved. However, the 2 conditions can usually be distinguished based on the identification of a nasogastric tube in an intrathoracic stomach or by instilling contrast and obtaining a limited study of the gastrointestinal tract. If a CCAM diagnosed antenatally is not seen on postnatal chest radiograph, a follow-up CT scan should be done, although certainly this is not required in the immediate postnatal period.
Infants with CCAMs who are symptomatic in the newborn period should undergo urgent surgical treatment, consisting of thoracotomy and either lobectomy or occasionally segmental resection if the lesion is amenable to this. The management of CCAMs that are asymptomatic at birth remains somewhat controversial, with some authors recommending observation and others advocating planned, elective excision. The most common complication of CCAMs that are asymptomatic at birth are the development of symptoms from air or fluid trapping and the development of infection in the cyst, which presents as acute-onset respiratory distress or pneumonia and is estimated to occur in at least 10% of patients who are initially asymptomatic.31
The other important consideration in the management of asymptomatic cystic lung lesions is the risk of malignancy. Malignant processes have historically been described in association with CPAM types 1 and 4: PPB in CPAM types 1 and 4 and much more rarely BAC in CPAM type 1. It should be restated that cystic PPB is not a preexisting CPAM that has undergone malignant transformation; rather, cystic PPB is a discrete low-grade tumor that evolves over 2–4 years to a high-grade, solid, overtly sarcomatous disease. Patients with PPB may have associated tumors, most commonly cystic nephroma.18 Over 90% of patients with PPB present before age 6, and the youngest patient to be diagnosed with PPB was only 2 months old.32
On the basis of the risks of infection and malignancy, the very low morbidity of infant thoracotomy, and the potential for compensatory lung growth up to approximately age 7, most pediatric surgeons favor elective resection of CCAMs after approximately 3 months of age.
BRONCHOPULMONARY SEQUESTRATION
Bronchopulmonary sequestrations are bronchopulmonary foregut malformations consisting of lung tissue that lacks connection to the tracheobronchial tree and account for 20% to 40% of congenital lung anomalies.33 Sequestrations may be invested by their own visceral pleura (extralobar) or are located within normal lung (intralobar), with the 2 forms occurring with approximately equal incidence. Extralobar sequestrations can be found above, below, and even within the diaphragm (Figure 28-3). Another characteristic feature of sequestrations is a systemic arterial supply, which typically arises from the descending thoracic aorta and occasionally the infradiaphragmatic aorta. Venous drainage may be either systemic (extralobar) or through the appropriate pulmonary vein (intralobar).
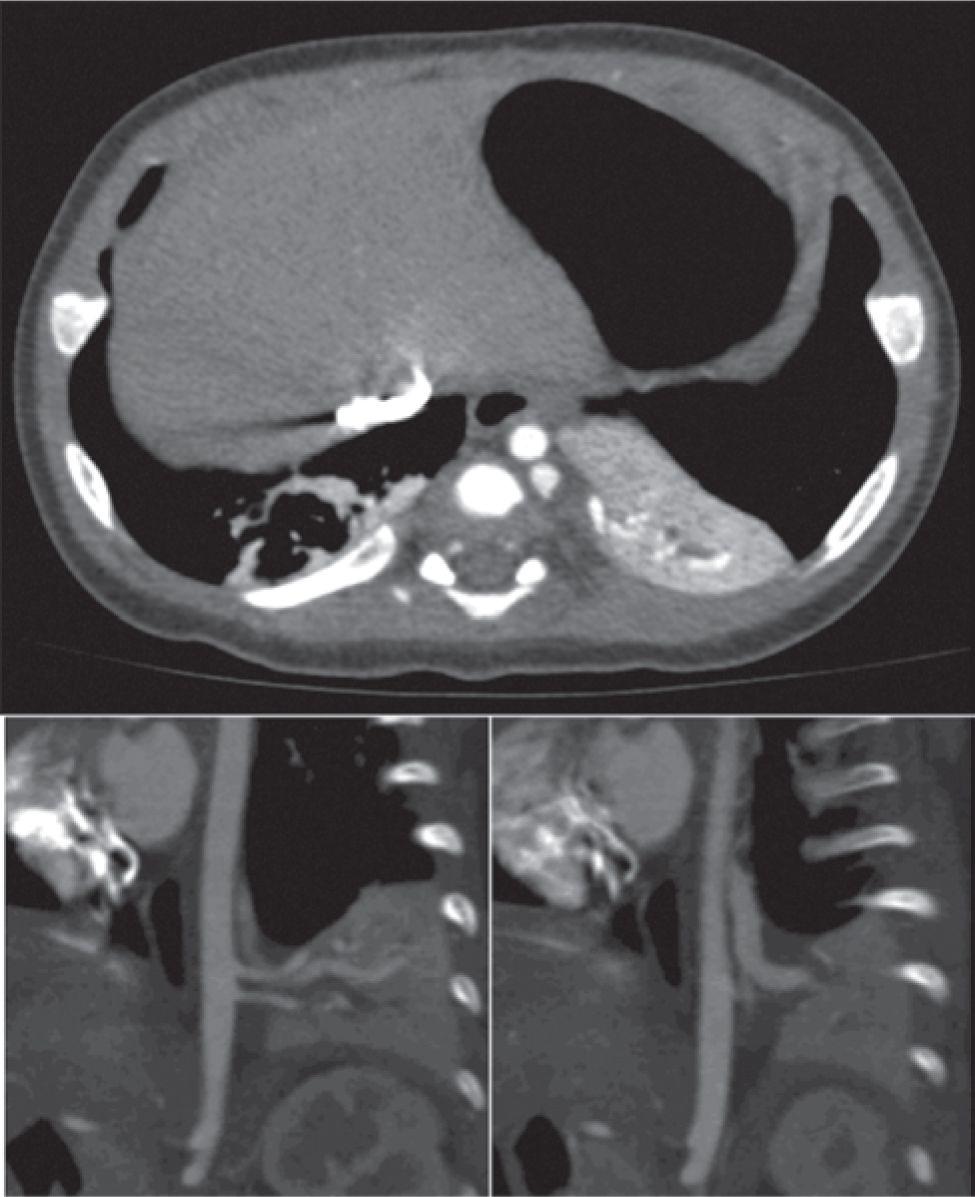
FIGURE 28-3 Computed tomographic scan demonstrating extralobar bronchopulmonary sequestration (BPS) located between left lower lobe and diaphragm. A, Axial view through BPS; B and C, left parasagittal views demonstrating two systemic arteries arising from the descending aorta (B) and systemic venous drainage into the hemiazygous vein (C).
Given their origin from foregut, there may occasionally be a connection between the sequestration
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


