Cystic Fibrosis
David M. Orenstein
 EPIDEMIOLOGY
EPIDEMIOLOGY
Cystic fibrosis (CF) is inherited as an autosomal recessive disorder. It is most common in those of northern European descent, with an incidence of approximately 1 in every 3200 live births. It is seen in about 1 in every 17,000 births in African Americans and is much less common in Asian populations. It has been found in virtually every ethnic and racial group. Approximately 4% of whites are heterozygous for CF (ie, carry one CF allele); heterozygotes have no evidence of clinical disease.
 PATHOPHYSIOLOGY AND GENETICS
PATHOPHYSIOLOGY AND GENETICS
CF affects virtually every organ system with epithelial surfaces—most importantly, the lungs, pancreas, intestinal mucus glands, liver, the reproductive tracts, and sweat glands. A common pathogenetic mechanism in major target systems is abnormal ion transport across epithelial surfaces. Impermeable chloride channels and over-active sodium pumps of these epithelial cells lead to biochemical and bioelectric abnormalities within organ lumina, leading in turn to viscid intralumenal secretions in the affected organs. These abnormally viscous secretions cause the blockage of ducts and air passages.
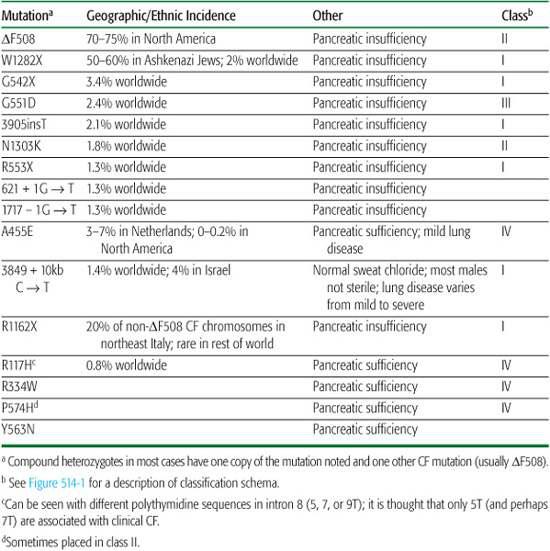
The most common mutation in the CF gene is a three-base-pair deletion that leads to the loss of a single phenylalanine at position 508 of the protein product (“ΔF508”).4 The ΔF508 mutation accounts for 70% to 80% of CF chromosomes; about 50% of CF patients in North America are homozygous for this mutation. More than 1500 other mutations at the CF locus have been discovered, but these account for only a small percentage of CF cases. In a few ethnic groups, a small number of mutations account for a large proportion of CF cases (Table 514-1). Prenatal testing and carrier testing can be accomplished in virtually every family desiring this information.
Table 514-1. Characteristics of Various CFTR Mutations
The gene for CF is on the long arm of chromosome 7 and spans 250 kb. Its product is a 1480–amino acid protein that regulates transmembrane ion transport.4 It serves as the most important apical membrane chloride channel and influences sodium and water transport across epithelial cells of many organs and glands.2,3 Because of this functional role, both the protein product and the gene itself are called cystic fibrosis transmembrane conductance regulator (CFTR). The gene is transcribed into mRNA, which is translated into protein in the endoplasmic reticulum (Fig. 514-1). Then, the CFTR protein is glycosylated in the Golgi apparatus and folded into a configuration that allows it to travel through the cytoplasm to the apical surface of epithelial cells. Once it resides in the apical membrane, it must have appropriate regulation, in that it must be able to respond to regulatory molecules. Finally, normal conductance of chloride and other ions depends on the channel remaining open for the appropriate time. CFTR mutations can be classified according to which step in this sequence of events is defective (Fig. 514-1).
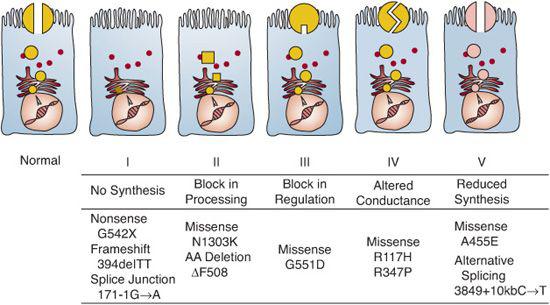
FIGURE 514-1. Molecular consequences of cystic fibrosis transmembrane conductance regulator (CFTR) mutations.
The ion transport abnormalities result in diminished amounts of airway surface liquid and therefore interfere with clearance of airway mucus, resulting in the airway problems common to patients with CF.3 There does seem to be a genotype-phenotype correlation for pancreatic status, with most CF genotypes, notably homozygosity for the ΔF508 mutation, associated with pancreatic insufficiency and a few alleles apparently conferring pancreatic sufficiency. Very few CF alleles appear to be associated with a delayed onset and slower progression of pulmonary disease, but by and large, genotype-phenotype relations for CF lung disease and survival have eluded discovery.2 Characteristics of some of the most common CFTR mutations are listed in Table 514-1. Because there is substantial phenotypic variation within the population of ΔF508 homozygotes, it is thought that there are genes other than CFTR that can modify the phenotypic expression of CF. Evidence for the existence of these modifier genes includes the observations that monozygotic twins have closer concordance of their clinical disease than do dizygotic twins. Variant alleles for both transforming growth factor β (TGF-β) and mannose binding lectin (MBL) have been associated with more rapid decline in lung function.5
 CLINICAL FEATURES
CLINICAL FEATURES
Gastrointestinal Tract
The typical cystic fibrosis (CF) patient has exocrine pancreatic insufficiency, with maldigestion of fats and protein and consequent malabsorption, steatorrhea, and failure to thrive.6 Pancreatic insufficiency is present at birth in 50% of CF patients and develops by age 9 years in another 35% to 40%.6 This is important to keep in mind, since the diagnosis of CF is often delayed or missed in patients without typical gastrointestinal involvement. Pancreatic status is determined in large part by genetic factors, as certain genotypes (eg, ΔF508) are nearly always associated with pancreatic insufficiency, while others confer pancreatic sufficiency (Table 514-1).
Bowel obstruction, a result of thickened intestinal mucus and pancreatic insufficiency, is present at birth (meconium ileus) in 10% to 20% of patients, especially those who are ΔF508 homozygotes. Later in life, distal intestinal obstruction syndrome (DIOS) occurs in 20% to 25% of affected individuals. Rectal prolapse, caused by bowel obstruction and by malnutrition with loss of anal sling musculature, is seen in 20% of CF patients in the first years of life. Intussusception is much less common, but CF patients account for a substantial portion of all patients with intussusception after 1 year of age. Gastroesophageal reflux, which may complicate the pulmonary disease, interfere with nutrition, or both, occurs with increased frequency in infants (and older children) with CF. Reflux may be worsened, especially in infants, by head-down positioning for chest physical therapy. Acid peptic disease can result from gastric acid hypersecretion and deficient pancreatic bicarbonate secretion. Cholelithiasis is more common in CF than healthy control populations. Liver pathology, including nonspecific steatosis; cholestasis; and the specific lesion, focal biliary cirrhosis, occurs in up to 10% of infants and children with CF. However, the clinical manifestation of cirrhosis with hepatic failure or portal hypertension with hypersplenism, bleeding esophageal varices, or both, is much less common. These hepatic complications present most commonly in the first decade and a half and seem not to be increasing in incidence as life span has increased. Children or adults, particularly those with normal exocrine pancreatic function, may develop recurrent episodes of acute pancreatitis. Endocrine pancreas dysfunction also can occur, leading to carbohydrate intolerance and diabetes mellitus that is unassociated with ketoacidosis.
Sweat Glands
The epithelial ion transport defect is expressed in the sweat glands, leading to the high salt content of CF sweat, long recognized as a hallmark of this disorder. In patients without CF, sweat precursor fluid is isotonic to plasma, and as the fluid moves through the sweat apparatus toward the skin, chloride is reabsorbed, with sodium following to maintain electrical neutrality. This results in sweat on the skin surface that is hypotonic to plasma and usually has sodium and chloride concentrations below 40 meq/L. In CF, the absent or poorly functioning cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel makes chloride reabsorption substantially below normal.2,7 The resultant fluid that emerges as sweat has sodium and chloride concentrations greater than 60 meq/L. This ion transport defect in the sweat apparatus provides the basis for the sweat test (discussed under “Diagnosis”). CF patients lose more salt during exercise in the heat and during febrile illnesses than do persons without CF, and they may experience dehydration or heat prostration. Infants may develop hyponatremia and hypochloremia. For unknown reasons, African American infants with CF seem to be at greater risk for this complication than Caucasian infants.6
Respiratory Tract
The upper respiratory tract is involved in virtually all cystic fibrosis (CF) patients, with radiographic evidence of pansinusitis. This is much more evident radiographically than symptomatically.6 It occasionally is helpful diagnostically (particularly in childhood), because persistent pansinusitis is uncommon except in CF, and CF is extremely uncommon without pansinusitis. Nasal polyps may be found in as many as 25% of CF patients and may be recurrent. Indeed, the finding of large nasal polyps in children less than 12 years of age should prompt the clinician to consider CF.
The lower respiratory tract involvement in CF accounts for over 90% of the morbidity and mortality. Although the lungs are histologically and radiographically normal at birth, the respiratory epithelium is abnormal electrophysiologically, leading to obstruction of small airways by viscid mucus, by airway inflammation, and by recurrent endobronchial infection, which typifies the disease.2,3 Invading organisms and inflammatory cells release inflammatory mediators and leave behind large amounts of DNA when the cells lyse, adding further to viscosity of lung mucus. Because of these factors, obstructive pulmonary disease, beginning in the small airways, eventually is present in almost all patients with CF. Recurrent cough, wheeze, or both, which may be diagnosed initially as recurrent bronchiolitis, asthma, or pneumonia, are often the first indications of pulmonary involvement. As the disease progresses, hyperinflation and crackles become apparent, and diffuse bronchiectasis eventually develops.
Pulmonary function tests reveal a pattern of obstructive airway disease: decreased forced expiratory volume in 1 second, decreased peak expiratory flow, and increased residual volume, indicative of air trapping. These obstructive changes show varying responses to bronchodilator inhalation: some patients improve, whereas many do not change, and a few actually worsen, because bronchiectatic airways may require bronchoconstrictor tone to remain stable. The response to bronchodilators is not consistent over time. Exercise testing6 typically shows reduced exercise tolerance and fitness, with relatively high minute ventilation for the oxygen consumed, presumably because of greater-than-normal dead-space ventilation. Often, a higher-than-normal proportion of the ventila-tory capacity is required at peak workloads. Male patients have greater exercise tolerance and cardiopulmonary fitness than female subjects. The pulmonary function and exercise tests are relatively sensitive tools for following progression of disease in the older, cooperative child (see Chapter 503).
Chronic pulmonary infection and inflammation, with episodes of acute exacerbation, are typical of CF patients.6,8 The chronic and acute conditions are most accurately thought of as purulent bronchiolitis and bronchitis. Exacerbations are characterized by increased cough, often producing purulent sputum, particularly in the morning on arising and with exertion; decreased exercise tolerance; lethargy; malaise; and weight loss. Fever is often absent. In the early stages of the disease, the bacterial organisms most commonly colonizing the lower respiratory tract of patients with CF include Staphylococcus aureus, Hemophilus influenzae, and a variety of gram-negative organisms. Eventually, most patients permanently acquire Pseudomonas aeruginosa and related gram-negative organisms, many of which become resistant to conventional antibiotic therapy. Increasing numbers of patients have Pseudomonas species at diagnosis; whether this represents a true increase in prevalence or represents better microbiology laboratory performance is uncertain. There seems to be a unique relationship between CF patients and Pseudomonas; at least half of all CF patients are colonized with a peculiar mucoid strain of this organism that is seldom seen in other human disease states. Some studies have suggested that early colonization of the respiratory tract with Pseudomonas is an independent risk factor for progressive pulmonary disease. Recently, other organisms, such as Aspergillus fumigatus, Burkholderia cepacia, Alcaligenes (Achromobacter) xylosoxidans, and Stenotrophomonas maltophilia, have become increasingly important as pulmonary pathogens. 
Progression of CF Lung Disease2,3,6,8
The rate of lung disease progression varies widely among individuals, influenced in part by environmental factors; secondhand cigarette smoke, lower socioeconomic status,9 and recurrent viral infection are three proven factors associated with worse pulmonary prognosis.
The initial histological lesion is bronchiolitis, reflected physiologically as small airways obstruction and radiographically as overinflation and prominent bronchial markings (eFig. 514.1  ). Further worsening of the lung disease includes extension from bronchiolitis to bronchitis, with thickened bronchial walls demonstrable on radiographs as circular lesions if the bronchi are projected in cross section or as characteristic parallel linear opacities (“tram tracks”) if the bronchi are projected longitudinally. With further progression and the development of bronchiectasis and small cysts, rounded densities become evident on radiographs. The right upper lobe is commonly affected earlier and more severely than other lobes (eFig. 514.2
). Further worsening of the lung disease includes extension from bronchiolitis to bronchitis, with thickened bronchial walls demonstrable on radiographs as circular lesions if the bronchi are projected in cross section or as characteristic parallel linear opacities (“tram tracks”) if the bronchi are projected longitudinally. With further progression and the development of bronchiectasis and small cysts, rounded densities become evident on radiographs. The right upper lobe is commonly affected earlier and more severely than other lobes (eFig. 514.2  ). With more advanced disease, small abnormal areas coalesce to form larger cysts (which may be dense when filled with mucopurulent secretions or may be lucent when relatively empty) and regions of fibrosis. Enlarged tortuous bronchial arteries may contribute to the opacities visible on chest radiographs and may also lead to hemoptysis. Large apical blebs may form and rupture, leading to pneumothorax (eFig. 514.3
). With more advanced disease, small abnormal areas coalesce to form larger cysts (which may be dense when filled with mucopurulent secretions or may be lucent when relatively empty) and regions of fibrosis. Enlarged tortuous bronchial arteries may contribute to the opacities visible on chest radiographs and may also lead to hemoptysis. Large apical blebs may form and rupture, leading to pneumothorax (eFig. 514.3  ).
).
Pulmonary complications include chest pain, pneumothorax, hemoptysis, segmental and lobar atelectasis, pulmonary hypertension leading to cor pulmonale, and respiratory failure (see the “Treatment of Pulmonary Complications” section). Digital clubbing is a nearly universal finding in patients with even mildly abnormal lung function.
Reproductive System
The reproductive tract is involved in most male patients, with atresia of the vas deferens and consequent obstructive azoospermia and sterility. However, male CF patients can produce children through in vitro methods. In female patients, thick cervical mucus often results in decreased fertility. Delayed puberty may be seen in either sex as a consequence of chronic illness and poor nutrition.
Impaired Glucose Tolerance and Diabetes
Some teens and adults with CF show an impaired insulin response, characterized initially by an impaired first-phase insulin response, then by a delayed and reduced peak insulin response. Decreased insulin sensitivity (insulin resistance) may also be present in patients with CF. Insulin is the only currently recommended therapy for all types of CF-related diabetes, and many clinicians find that basal/bolus regimens are optimal. Adolescents and adults display a unique pattern of hyperglycemia. Abnormal glucose tolerance tests are frequent, but diabetic ketoacidosis is rare, and microvascular complications of diabetes are less frequent. With improved survival, microvascular diabetic complications are likely to be seen more frequently.
Bone Complications
Patients with CF, especially those who are older, have a higher incidence of bone fracture caused by decreased bone density. This may be secondary to malabsorption of vitamin D and calcium, hypogonadism, inactivity, or cytokines from chronic infection. Contemporary management with appropriate vitamin and nutritional supplementation likely limits this complication. Exercise during childhood may be helpful. Occasional patients, particularly those with severe lung disease, have hyper-trophic pulmonary osteoarthropathy involving the long bones and adjacent joints. This complication is characterized clinically by joint (especially knee) pain and radiographically by periostial thickening. A systemic vasculitis syndrome with arthritis and a vasculitic skin rash has also been described in CF patients.
 DIAGNOSIS
DIAGNOSIS
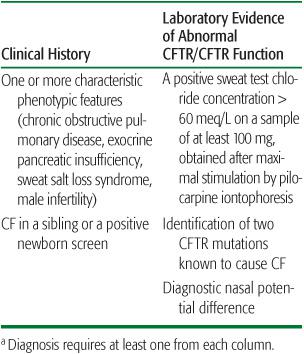
The key to diagnosing cystic fibrosis (CF) is a high index of suspicion in the presence of any of the manifestations. Increasingly, the diagnosis of CF is made by newborn screening, but diagnosis still requires confirmatory evaluation and testing in a specialized CF center. CF is rarely diagnosed without a confirmatory positive sweat test. The accepted criteria for diagnosis of CF are shown in Table 514-2.10
Newborn Screening
Most newborns with CF have an elevation of blood immunoreactive trypsinogen (IRT). The assay for IRT can be carried out from the dried blood spots obtained from newborns for routine screening for other genetic and metabolic diseases. The IRT test is used in more than 30 states and in many countries, and its use is likely to continue spreading. There appear to be very few false negatives with this screen, but the false-positive rate is as high as 90%. Some screening programs move directly to genetic analysis for the most common cystic fibrosis transmembrane conductance regulator (CFTR) mutations in blood spots with elevated IRT, while others repeat the analysis for IRT after one elevated level. If the second test still shows elevated IRT, or if one or two CFTR mutations are identified on DNA analysis, prompt referral to a CF center is indicated for definitive genetic or sweat testing and initiation of comprehensive treatment for those confirmed to have CF.
Table 514-2 Diagnostic Criteria for Cystic Fibrosisa
Sweat Testing
Sweat testing remains the gold standard for diagnosis of CF. Table 514-3 lists the indications for performing a sweat test. Most physicians are sufficiently aware of the disease, so few children with the triad of growth failure, steatorrhea, and chronic pulmonary disease escape diagnosis. However, atypical patients, especially those who have no clinically apparent pancreatic involvement (as many as 15% of all CF patients and as many as 50% of young infants with CF) or who have normal growth may escape diagnosis for years.
Theoretically, the sweat test is simple, but false positives and false negatives are extremely common in tests performed outside established CF centers.7 Contrary to widespread belief, sweat tests can be accomplished in young infants, although some young infants might not produce a large enough volume of sweat for analysis. Concentrations of sodium and chloride in sweat are below 40 meq/L in normals, but nearly all patients with CF have values greater than 60 meq/L. Very few patients fall in the intermediate or borderline range (40–60 meq/L); in these individuals, genotype analysis or measuring nasal potential difference may be required for definitive diagnosis. Patients with intact exocrine pancreatic function have somewhat lower sweat chloride concentrations than those with pancreatic insufficiency, but their values are still well outside the normal range. Table 514-4 lists conditions giving false-positive and false-negative sweat test results.
Nasal Potential Difference
The function of the CFTR protein in respiratory epithelium can also be assessed directly in vivo by measuring the bioelectric voltage difference across nasal epithelium (the “nasal potential difference,” or nasal PD); this is available in a few specialized CF centers.10
Table 514-3 Indications for Sweat Testing
Gastrointestinal tract |
|
|
|
|
|
|
|
|
Respiratory tract |
|
|
|
|
|
|
|
|
|
|
|
Other |
|
|
|
|
|
|
|



