Cystic Diseases of the Kidney
Friedhelm Hildebrandt
Virtually all renal cystic illnesses are monogenic diseases (Table 470-1). A recent unifying theory of their pathophysiology suggests that all gene products (“cystoproteins”) that are mutated in cystic kidney diseases are expressed in primary cilia, basal bodies, or centrosomes.1-4 Primary cilia are antennalike cellular organelles produced by virtually every epithelial cell type in the body. The structure and function of primary cilia and basal bodies is delineated in eFig. 470.1  .5,6 They are important for perceiving extracellular cues, including photosensation, mechanosensation, osmosensation, and olfactory sensation. Cilia are assembled from basal bodies, which represent one of the two centrosomes. Centrosomes and basal bodies contain the same protein complexes that are part of the mitotic spindle in mitosis. These protein complexes are crucial for planar cell polarity, or the orientation of epithelial cells in three-dimensional space. Disruption of their function leads to cyst development and to extrarenal defects that have been summarized under the term ciliopathies. In general, it seems that the pathogenesis of ciliopathies is based on an inability of epithelial cells to sense or process extracellular cues.7
.5,6 They are important for perceiving extracellular cues, including photosensation, mechanosensation, osmosensation, and olfactory sensation. Cilia are assembled from basal bodies, which represent one of the two centrosomes. Centrosomes and basal bodies contain the same protein complexes that are part of the mitotic spindle in mitosis. These protein complexes are crucial for planar cell polarity, or the orientation of epithelial cells in three-dimensional space. Disruption of their function leads to cyst development and to extrarenal defects that have been summarized under the term ciliopathies. In general, it seems that the pathogenesis of ciliopathies is based on an inability of epithelial cells to sense or process extracellular cues.7
Table 470-1. Cystic Renal Disorders
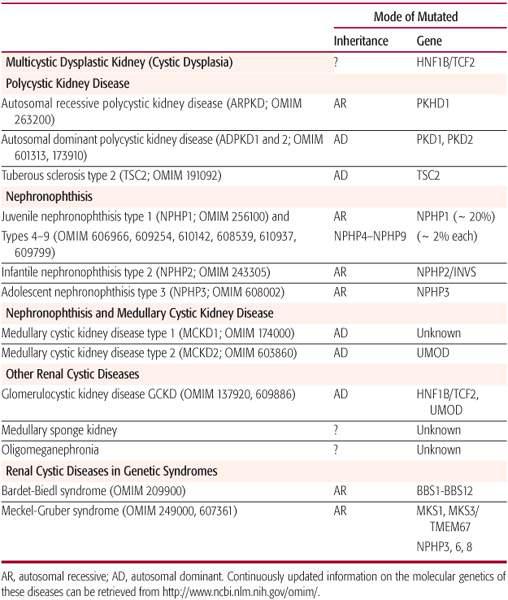
DIAGNOSIS
Cystic disorders of the kidney are among the most common causes for end-stage kidney disease (ESKD) in children. Since most renal cystic diseases are monogenic disorders, pedigree analysis to determine the pattern of inheritance should be emphasized when obtaining the history. In autosomal dominant diseases, a genetic defect in only one of the two alleles of the disease gene leads to the disease phenotype. Typically, there are affected individuals in more than one generation. Father-to-son transmission may be present (as opposed to X-linked diseases), and the disease course is usually milder and has later onset than in recessive variants of the disease. In autosomal recessive diseases, defects in both alleles of a gene (on both parental chromosomes) are necessary for the disease to occur. Typically, there are affected individuals in only one generation, the disease course is usually more severe than in a dominant variant of the condition, and onset occurs already in childhood and adolescence. Knowing the mode of inheritance is an important guide for differential diagnosis, for genetic counseling, and for obtaining a molecular genetic diagnosis. In some diseases, defects in different genes localized at different chromosomal loci can give rise to identical or very similar disease phenotypes in different patients. This phenomenon is called genetic locus heterogeneity and is exemplified by autosomal dominant polycystic kidney disease (ADPKD) types 1 and 2.
In some renal cystic disorders, the responsible gene has been identified, so a definitive diagnosis may be made by direct molecular genetic testing that identifies the disease-causing mutation in affected patients. (Updates on laboratories that perform molecular genetic diagnoses can be found at http://www.genetests.org/ and www.genetests.org/. renalgenes.org.) Molecular genetic diagnosis should be initiated only within the context of genetic counseling by a certified genetic counselor, because there are many difficult ethical issues to consider.
All renal cystic disorders manifest with characteristic changes on ultrasound examination and present at specific age ranges. Therefore, an algorithm for differential diagnosis in cystic diseases of the kidney is based on results of renal ultrasound and is stratified by certain age groups and on molecular genetic diagnostics. A diagnostic algorithm for renal cystic disease is shown in Figure 470-1.
MANAGEMENT
Renal cystic disorders eventually lead to end-stage kidney disease (ESKD). No treatment is available that changes disease progression or treats the underlying cause of these disorders. Therefore, therapy is symptomatic and is aimed at complications that might accelerate progression. This includes controlling blood pressure in patients with autosomal dominant polycystic kidney disease (ADPKD) to decelerate progression into renal failure. In the phase of compensated chronic renal insufficiency, symptomatic treatment for anemia, acidosis, growth retardation, and renal osteodystrophy is most important (see Chapter 477). Eventually, renal replacement therapy by dialysis and transplantation becomes necessary (see Chapters 129 and 478). Recently, it was shown that renal cystic mouse models are responsive to treatment with a vasopressor type 2 antagonist,8,9 with everolimus, or with roscovitine.10 Phase 2 treatment trials for cystic kidney disease in adults have been initiated with some of these agents.
MULTICYSTIC DYSPLASTIC KIDNEY (MCDK)
MCDK, also termed cystic dysplasia, is a developmental defect in kidney organogenesis that is discussed in Chapter 469.
AUTOSOMAL RECESSIVE POLYCYSTIC KIDNEY DISEASE
Autosomal recessive polycystic kidney disease (ARPKD) occurs in 1 in 6000 to 40,000 live births. In a study of 66 boys and 49 girls with ARPKD, age at diagnosis was 11% prenatal, 41% neonatal, 23% infantile, and 25% juvenile (Fig. 470-2). Uniformly, there is bilateral kidney enlargement as a result of transformation of collecting ducts into fusiform cysts. Among extrarenal organ manifestations, congenital hepatic fibrosis is the most important one. ARPKD is always associated with hepatic fibrosis, which may not be clinically evident initially, because renal failure frequently develops earlier than clinical sequelae of hepatic fibrosis. Over time, cystic dilation of the biliary tree (Caroli disease) develops, followed by portal hypertension with a risk of GI bleeding from esophageal varices. Liver cell function is initially normal until cirrhosis develops. Rarely, cysts are present in the pancreas or spleen. A few patients will not manifest the renal components of their disease and will have reasonably well-preserved renal function but will present with symptoms related to congenital hepatic fibrosis and portal hypertension.
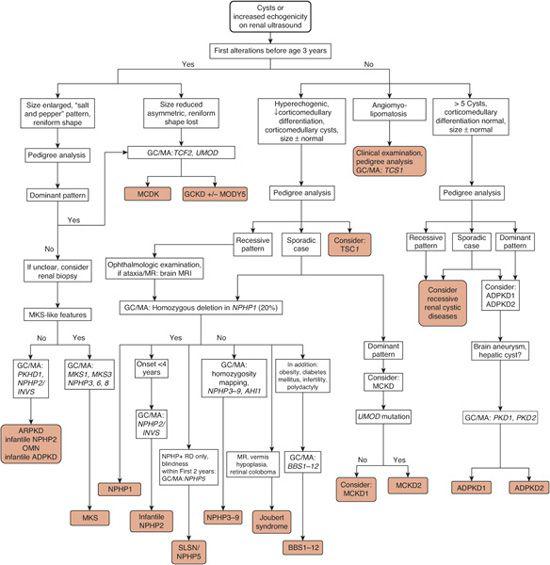
FIGURE 470-1. Algorithm for the evaluation of renal cystic disorders based on renal ultrasound and clinical and molecular genetics. Resultant diagnoses are shown on gray background. GC/MA, initiate genetic counseling (GC) and perform mutation analysis (MA) of the gene(s) indicated in italics (http://www.renalgenes.org; www.genetests.org). ADPKD, autosomal dominant polycystic kidney disease; infantile ADPKD, ADPKD due to homozygous deletions; ARPKD, autosomal recessive polycystic kidney disease; BBS, Bardet-Biedl syndrome; GCKD, glomerulocystic kidney disease; MCKD, medullary cystic kidney disease; MKS, Meckel-Gruber syndrome; MODYS, maturity-onset diabetes of the young; MR, mental retardation; NPHP1, 2, etc, nephronophthisis types 1, 2, etc, respectively; OMN, oligomeganephronia; SLSN, Senior-Løken syndrome.
A single recessive gene, PKHD1, has been identified by positional cloning as causing ARPDK. Because ARPKD is a recessive disease, an a priori risk of 25% exists for a sibling of an affected individual. Autosomal recessive polycystic kidney disease carries a high lethality in the perinatal period if two truncating mutations are present in the PKHD1 gene. If at least one missense mutation is present, end stage renal disease (ESRD) develops later in childhood or adolescence.12
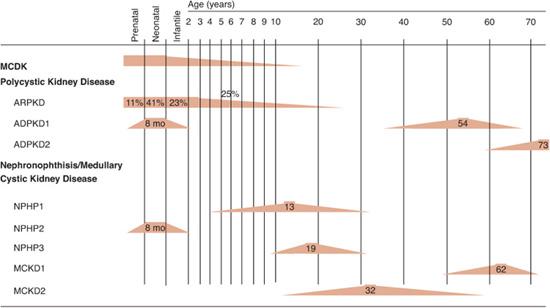
FIGURE 470-2. Time course of renal failure in renal cystic disorders. Range for age of onset of end-stage renal disease is given as color rectangular symbols. Range for age at diagnosis is shown as color rectangular symbols. Numbers indicate median age in years. ARPKD, autosomal recessive polycystic kidney disease. ADPKD, autosomal dominant polycystic kidney disease. BBS, Bardet-Biedl syndrome; GCKD, familial hypoplastic glomerulocystic kidney disease; NPHP, nephronophthisis; MCDK, multicystic dysplastic kidney; MCKD, medullary cystic kidney disease.
 DIAGNOSIS
DIAGNOSIS
Diagnosis of ARPKD rests on clinical signs and symptoms, renal and hepatic imaging, and molecular genetic diagnostics, as outlined in Figure 470-1. Prenatal findings in ARPKD include oligohydramnios and a renal sonographic echotexture known as salt-and-pepper appearance. Enlargement of fetal kidneys may not occur until after 24 to 28 weeks of gestation, making early diagnosis difficult. Oligohydramnios from intrauterine renal failure is associated with insufficient lung development and the typical “Potter facies,” which is characterized by low-set ears, a flat nose, and a retracted chin. In these severe prenatal cases, abnormal kidney growth can already be detected by ultrasound in the second half of pregnancy. A definitive diagnosis of ARPKD is theoretically possible by direct mutation analysis of the PKHD1 gene,12 with an approximate 80% detection rate. Mutation analysis of the PKHD1 gene should be initiated with the guidance of a genetic counselor, for both pre- and postnatal testing. For the diagnosis of ARPKD, a negative renal ultrasound in the parents is obligatory in order to distinguish the disease from early-onset autosomal dominant polycystic kidney disease (ADPKD).
Initial symptoms and signs that lead to the diagnosis of autosomal recessive polycystic kidney disease (ARPKD) in infants and children include a palpable abdominal mass, hypertension, urinary tract infections, an affected sibling, liver involvement, and growth failure. Bilaterally enlarged hyperechogenic kidneys maintain a reniform shape even when grossly enlarged. On ultrasound, kidneys continue to exhibit a salt-and-pepper appearance until later stages, when distinct, rounded cysts appear. In ARPKD, kidneys can be grossly enlarged so that palpable abdominal masses are common, as are hypertension and urinary tract infections. On hepatic ultrasound, the liver is hyperechogenic, and later visible biliary cysts may develop. Renal and hepatic changes can also be confirmed by magnetic resonance imaging, where the radial arrangement of fusiformly dilated collecting ducts can be noted. Kidney biopsy plays a decreasing role in diagnosis due to molecular genetic diagnostics. On renal histology, there is fusiform dilation and cyst development, involving 10% to 90% of collecting ducts, depending on severity and age of onset. Liver histology exhibits portal and interlobular fibrosis as well as biliary duct hyperplasia. Liver biopsy is useful to assess the degree of liver failure in later stages.
 DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
Other cystic diseases that occur in infancy and early childhood need to be considered (Fig. 470-3). Specifically, medullary cystic kidney disease, infantile nephronophthisis, and Meckel-Gruber syndrome can be confused with ARPKD (Table 470-1). Multicystic dysplastic kidney can be distinguished from ARPKD by reduced kidney size and loss of the reniform shape. Autosomal dominant polycystic kidney disease (ADPKD) may occur rarely in infants if large deletions are present in the PKD1 gene. Meckel-Gruber syndrome, in addition to renal agenesis and dysgenesis shows a wide variety of extrarenal organ involvement, including occipital encephalocele, cleft palate, polydactyly, and ocular anomalies and usually leads to perinatal lethality. A rare but important differential diagnosis is infantile NPHP, which can be diagnosed by mutation analysis of the NPHP2/INVS gene (www.renalgenes.org). Bilateral Wilms tumor is rare and can be distinguished from ARPKD by a highly irregular parenchymal pattern on imaging techniques. Bardet-Biedl syndrome can clearly be distinguished from ARPKD by the presence of retinitis pigmentosa and polydactyly, together with obesity, diabetes mellitus, mental retardation, and hypogonadism.
 COMPLICATIONS
COMPLICATIONS
Chronic renal failure with anemia, growth retardation, and osteodystrophy is common. There is a high risk of urinary tract infection. Urinary outflow obstruction from renal cysts may also occur. Hypertension is frequent, and nephrolithiasis occurs in about 15% of cases. Esophageal variceal bleeding and hyper-splenism can result from liver fibrosis. Ascending cholangitis occurs from infections of hepatic cysts. Fertility- and pregnancy-related problems are also common.
 TREATMENT
TREATMENT
The severe hypertension that occurs in young infants with ARPKD requires aggressive management to optimally preserve renal function. Early detection and aggressive treatment of urinary tract infections is also important. Managing portal hypertension frequently becomes a major problem. In end-stage renal disease, renal replacement therapy by dialysis and transplantation becomes necessary. Depending on whether liver failure has developed, combined kidney and liver transplantation or secondary liver transplantation after initial kidney transplantation may be warranted. Some experimental treatment forms for ADPKD are being developed in mouse models (see above; http://www.nih.gov/news/pr/jan2006/niddk-24.htm).
 PROGNOSIS
PROGNOSIS
Children presenting perinatally have a 30% to 50% mortality rate in the first month of life.12 Those who survive through the first month have a 1-year survival rate of 85% and a 10-year survival rate of 82%. Chronic renal failure is first detected at a mean age of 4 years. First renal replacement therapy was necessary by 5 years of age in 86% of cases. Three quarters of cases developed systemic hypertension. Sequelae of congenital hepatic fibrosis and portal hypertension developed in 45% of patients and were related to age.
AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE
Autosomal dominant polycystic kidney disease (ADPKD) is the most frequent lethal disease of autosomal dominant inheritance in the United States and Europe,13 with a prevalence of 1:1000 in the general population. About 10% to 15% of all patients in adult chronic dialysis programs have ADPKD. ESKD usually develops between age 60 and 70 years. Clinically, ADPKD2 is very similar to ADPKD1 but follows a somewhat milder course toward renal failure and does not present in childhood. ADPKD usually manifests in late adulthood but can lead to clinical symptoms in older children and young adults, including abdominal pain, palpable abdominal mass, hematuria, urinary tract infections, hypertension, and abdominal or inguinal hernias. Cerebral aneurysms and diverticulosis of the gut may develop in adults with ADPKD.
The PKD1 gene is localized on chromosome 16p13.1 and codes for polycystin-1, a protein with a large transmembrane domain and a complex extracellular domain. Polycystin-1 interacts with the gene product (polycystin-2) of the PKD2 gene. Although a germ-line mutation segregates in affected families in an autosomal dominant fashion, the disease mechanism, by which renal cysts develop gradually over several decades, is due to the loss of heterozygosity (LOH) of the second allele in individual cells, consistent with a “second hit mechanism” (see Chapter 170). In rare instances, ADPKD can occur in infancy if patients carry large deletions, which affect the PKD1 gene and the neighboring TSC2 gene, in way of a “contiguous gene deletion syndrome” (see Chapter 170). Among patients with tuberous sclerosis, apparently only individuals carrying such extensive deletions develop large bilateral renal cysts and exhibit infantile onset of ADPKD.14
 DIAGNOSIS
DIAGNOSIS
In children who are known to be genetically at risk due to ADPKD in a parent, the presence of even a single cyst in normal-sized kidneys is highly predictive for the development of ADPKD. Hepatic, pancreatic, or ovarian cysts are rarely detected before puberty. A diagnostic algorithm based on renal ultrasound and mutation analysis is shown in Figure 470-1. Molecular genetic diagnostics of PKD1 can be initiated after genetic counseling. Early genetic diagnosis may be valuable to monitor for complications such as hypertension, UTI, and cerebral aneurysms and to evaluate living related kidney transplant donors.
 TREATMENT
TREATMENT
Although in ADPKD chronic renal failure develops at a median age of approximately 65 years, at-risk children and young adults should be followed annually for hematuria and hypertension, and at greater intervals by renal ultrasound, especially during pregnancy. Aggressive treatment of hypertension or urinary tract infection is indicated, because both pose considerable risk for accelerated progression into ESKD. In families with a history of cerebral aneurysms, or if symptoms warrant, cranial CT or MRI should be followed, but routine screening is not usually undertaken. Phase 2 trials have been initiated for some experimental treatment approaches in adult patients with cystic kidney diseases.
 NEPHRONOPHTHISIS AND MEDULLARY CYSTIC KIDNEY DISEASE
NEPHRONOPHTHISIS AND MEDULLARY CYSTIC KIDNEY DISEASE
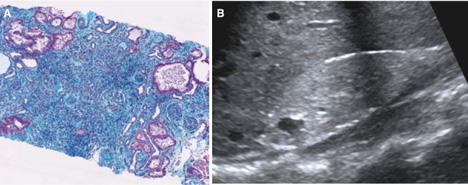
Diseases of the “nephronophthisis/medullary cystic kidney disease (NPHP/MCKD) complex” are renal cystic diseases that share a virtually identical renal histology15 characterized by thickening and disintegration of the tubular basement membrane, interstitial lymphocytic infiltrations with fibrosis, and distal tubular atrophy with cysts (Fig. 470-3A). Over time, chronic sclerosing tubulointerstitial nephropathy develops. In contrast to polycystic kidney disease, cysts occur primarily at the corticomedullary junction of the kidneys, and kidney size remains normal or is slightly diminished (Fig. 470-3B). Although histology is similar in all forms of the disease, the two different disease groups of recessive nephronophthisis (NPHP) and dominant medullary cystic kidney disease (MCKD) can be clearly distinguished by age of onset or pattern of inheritance.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


