Fig. 16.1
Preoperative coronal (a) and sagittal (b) T1-weighted MR images following gadolinium infusion showing a predominantly cystic sellar and suprasellar craniopharyngioma in a 2 year old. Postoperative coronal (c) images showing a significant reduction in the size of the cyst after image-guided drainage and placement of an Ommaya reservoir
Cystic contents are often proteinaceous and may appear as hyperintense on T1- and T2-weighted MRI compared to CSF. The solid tumor itself is hypo- to isointense on T1-weighted and heterogeneously hyperintense on T2-weighted MRI. There is typically heterogeneous enhancement of the solid component and often smooth enhancement of the cyst wall.
Neuroimaging must also characterize the degree and nature of hydrocephalus, if it is present, and is essential for planning operative approaches. Many centers use image navigation and intraoperative imaging as useful adjuvants for surgical management. Follow-up protocols have not been formally studied and are often based on personal or institutional preferences. Few case reports of antenatal diagnosis by ultrasound are available. Craniopharyngiomas may be discovered as incidental findings on neuroimaging for other indications.
While MRI and CT are quite characteristic for craniopharyngioma, there are some similar-appearing lesions, such as Rathke’s cleft cyst, xanthogranuloma, dermoid or epidermoid cysts, germ cell tumors, and low-grade glioma, or some pituitary adenomas (potentially with calcified hemorrhagic infarction) that should be considered in the differential diagnosis.
Pathology/Biology
The anterior pituitary gland is formed during gestation by the infolding of nasopharyngeal cells. Remnants of these cells are thought to give rise to some pineal region tumors and cysts in addition to craniopharyngiomas. This pedigree explains their resemblance to adamantinoma (enamel-forming tumor) of the jaw and calcifying odontogenic cysts. This origin also explains how craniopharyngiomas can also appear in the nasopharynx or sphenoid bone, as these are along the path of the infolding cells. Few craniopharyngiomas arise solely in the third ventricle, putatively growing from rest cells (remnants of the infolding) in the tuber cinereum. An alternate theory of origin attributes craniopharyngiomas to metaplasia of pituitary adenoid cells.
There are two variants of craniopharyngioma, adamantinomatous and papillary, though transitional types cloud this distinction. Papillary craniopharyngiomas occur nearly exclusively in adults, can be easier to resect, recur less often, and calcify and form cysts less often. Both subtypes are histologically benign tumors and only exceptionally show malignant transformation or CNS seeding. The following description focuses on the pediatric, adamantinomatous type of craniopharyngioma.
Macroscopically, the majority of pediatric craniopharyngiomas are multilobular cystic tumor with or without a solid component. The cysts are filled with thick, gritty, “machine-oil” fluid brownish in appearance. It is this fluid that can cause an aseptic meningitis if spillage occurs. The walls of the cyst may have a smooth lining and can be calcified leading to an eggshell appearance. The solid portion of the tumor is hard and gray and often has calcifications throughout making it crumbly. The tumor may be adherent to brain parenchyma, infundibulum, or optic pathway and may invaginate the third ventricle/foramen of Munro region.
Microscopically, craniopharyngiomas are microcystic, with cysts lined by squamous epithelial cells (Fig. 16.2). There may be a peripheral rim of palisading columnar cells. Sheets of cells keratinize and form anuclear ghost cells, creating “wet keratin.” Areas of dystrophic calcification give the tumor its adamantinomatous name. Tumor cells may secrete fluid and cholesterol, forming clefts and necrotic debris, or the fluid may be retained in the cells creating a looser matrix in regions.
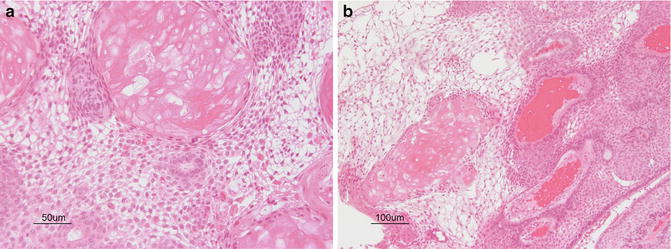
Fig. 16.2
(a, b) Cystic spaces are lined by neoplastic cells arranged in palisaded profiles. Cuboidal to columnar epithelium blends into looser central zones containing stellate cells. Calcification and clumps of “wet keratin” consisting of ghosts of keratinized cells are a prominent feature of the adamantinomatous variant of craniopharyngioma. Courtesy of Cynthia E. Hawkins
Tumor may interdigitate into the parenchyma without infiltration or causing frank invasion. The arachnoid may be absent in areas of significant gliosis or interdigitation.
Multinucleated giant cells may be present surrounding necrotic debris and cholesterol clefts; however, these are more typical for xanthogranuloma. The surrounding brain may show gliotic changes with Rosenthal fibers as a reactive process owing to the slow growth of these tumors. This should not be mistaken for pilocytic astrocytoma. Epidermoid and Rathke’s cleft cyst may be raised in the differential diagnosis but should be distinguishable by the experienced neuropathologist. As the majority of adamantinomatous craniopharyngioma harbors a mutation of the β-catenin gene, nuclear accumulation of β-catenin can aid in the distinction.
Current Treatment
Optimal management of craniopharyngiomas remains one of the most hotly debated topics in pediatric neurosurgery, neuro-oncology, and endocrinology. While total resection may confer the best long-term prospect for cure, patients can be left with devastating hypothalamic and pituitary dysfunction [8]. Neurosurgeons have attempted to reduce patient morbidity using microsurgical techniques, transnasal, transventricular, or a variety of skull-base approaches with the assistance of newer technology such as endoscopy, intraoperative MRI and ultrasound, and image-guided navigation, all with varying degrees of success. Alternative approaches, aimed at protecting the visual system and treating hydrocephalus, have therefore been pioneered with resultant improvements in quality of life, but higher progression rates. These approaches include subtotal resection, cyst drainage including placement of Ommaya reservoirs, intracystic chemotherapy, and radiotherapy. Prospective data with sufficient patient numbers to support a specific management approach is limited.
Surgical
Neurosurgeons who manage craniopharyngiomas have a number of choices available to them. Firstly, the extent of resection must be decided upon. The UK Children’s Cancer and Leukaemia Group consensus guidelines recommend total resection for tumors smaller than 2–4 cm without hypothalamic involvement or hydrocephalus in order to minimize postoperative worsening of pituitary, hypothalamic, and visual function [9]. Larger tumors, and those with hypothalamic involvement or hydrocephalus, are preferentially treated by subtotal resection followed by radiotherapy (according to age). Both of these approaches must be customized to the individual patient.
Upon deciding to proceed with surgery, multiple approaches are available. Transsphenoidal and endoscopic/endonasal surgeries approach the tumor through the sphenoid sinus and sella and may be extended through the diaphragm, planum sphenoidale, or the dorsum sella. It provides limited exposure to lateral and intraventricular tumor lobules, but may result in shorter hospital stays [10] (Fig. 16.3). Transcallosal or transventricular approaches are used to debulk tumors with extensive protrusion into the third ventricle; many centers use endoscopes to assist with these approaches. The limitation of a superior approach is access to infra-chiasmatic structures. Skull-base approaches provide multiple avenues for supra- and intrasellar tumors, with options to use pterional, subfrontal, frontolateral, or extended approaches for resection. Lateral approaches may not provide enough exposure of both optic nerves and intraventricular tumor lobules, and subfrontal (particularly bifrontal) approaches risk injury to both olfactory nerves and both inferior frontal lobes and have limited access to retrosellar regions and CSF cisterns. Ultimately, tumors with intrasellar, suprasellar, and/or intraventricular growth patterns may require combined approaches, capitalizing on the strengths of each in a staged or simultaneous manner.
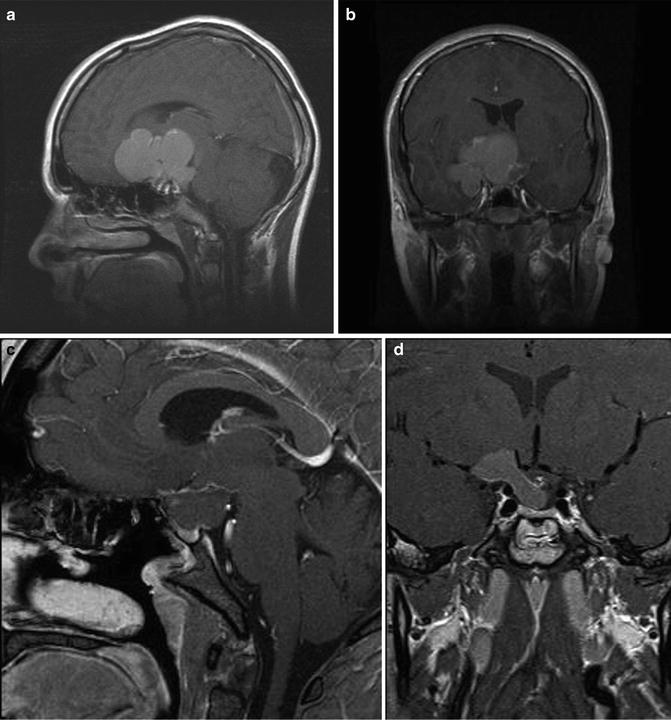
Fig. 16.3
Preoperative sagittal (a) and coronal (b) T1-weighted MR images following administration of gadolinium showing a multilobulated cystic craniopharyngioma in a 13 year old. Postoperative sagittal (c) and coronal (d) images showing reduction in the size of the cyst after endoscopic endonasal drainage
Since craniopharyngiomas grow superiorly, they should have arachnoid dissection planes with overlying structures. Unfortunately, this is not always the case. Many will induce a gliotic rim that can provide a pseudo-dissection plane. Care must be taken to preserve the infundibulum, optic apparatus, and hypothalamus. As well, perforating arteries and superior hypophyseal arteries (supplying the optic chiasm) may be stretched and displaced superolaterally and should be preserved. Dissection may be simpler prior to cyst drainage. Gentle retraction of the capsule as opposed to the nervous tissue is essential. Exposure may be improved by drilling off the planum sphenoidale and/or optic strut. Entry into the frontal or sphenoid sinuses requires careful closure or exenteration.
Often a large cyst may be aspirated prior to or instead of surgical resection. Cyst aspiration may be done under direct visualization, endoscopically, transsphenoidally, or via stereotactic needle or drainage tube placement. Draining a cyst early may expose the dissection plane better or may be a sufficient treatment for hydrocephalus. Care must be taken to avoid spillage of the cyst contents to avoid aseptic meningitis. For cysts that will require repeated aspirations, one may insert a subcutaneous catheter and reservoir, such as an Ommaya or Rickham reservoir, and/or use intracavitary sclerosing therapy (see below). Prior to intracystic chemotherapy, most institutional protocols require instillation of contrast medium to ensure the agent does not leak out of the cyst.
Hydrocephalus adds an extra dimension to surgical management of craniopharyngiomas, often requiring a first operation to divert CSF or drain an enlarged cyst. Acute hydrocephalus is typically treated with an extraventricular drain, while subacute presentations may be treated with endoscopic or stereotactic cyst aspiration. Septal perforation is occasionally beneficial for significant involvement of the foramen of Munro. CSF shunting may be required for a subset of patients, though most surgeons will initially attempt to reestablish CSF drainage pathways prior to committing a patient to a shunt.
Surgical complications are unfortunately common in these complex tumors. The most common complication, as mentioned previously, is pituitary injury causing transient or permanent hypopituitarism and diabetes insipidus. Hypothalamic injury is more common in patients with preoperative symptoms. Visual deterioration may occur due to manipulation of the optic chiasm or its vascular supply. Aseptic meningitis results from cyst content leakage and presents immediately, while septic meningitis takes a few days to develop. Communicating hydrocephalus can be a further complication of meningitis or occur despite reestablishment of the normal CSF pathways. Postoperative CSF leaks may be a sign of hydrocephalus or due to penetration of the sphenoid sinus, frontal sinus, or ventricular system; they may respond to temporary drainage via EVD or lumbar drain. Lastly, injury to the nerves or anterior circulation vascular structures may cause other focal neurological deficits, with anosmia and ageusia being common after subfrontal approaches.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


