Cranial Developmental Abnormalities
Omar Khwaja
The central nervous system (CNS) is the most complex organ in the human body comprising a highly organized anatomic scaffold of billions of cells and a network of trillions of connections. Structural brain development proceeds through an integrated series of often rapid developmental events from early embryogenesis through fetal life and into early adulthood. It is therefore not surprising that many common childhood neurologic and developmental disorders have their origins in genetic or environmental perturbations of embryonic or fetal brain development.
Advances in genetics and diagnostic imaging, including prenatal imaging, have led to earlier and more complete diagnosis and enhanced prognostication for CNS malformations and the ability to counsel effectively regarding recurrence. The diagnosis of malformations has been revolutionized by magnetic resonance imaging (MRI), and the challenge for the pediatrician is often to translate the imaging diagnosis to effectively counsel the family and plan further care and management for the child. Modern neonatal neurologic and neurosurgical intensive care together with progress in early intervention and pediatric rehabilitation means children with malformations of the brain or spinal cord may now survive longer and with improved quality of life.
Malformations of the CNS are among the most common problems in child neurology. Brain development is abnormal in an estimated 25% of conceptions and is responsible for a high percentage of miscarriage and stillbirth. Brain malformations are, together with congenital heart disease, the leading cause of neonatal and postneonatal mortality in the developed world. Brain malformations are after cerebral palsy, the leading cause of childhood morbidity and mortality related primarily to consequent neurologic disability and epilepsy. Brain malformations frequently coexist with other malformations of organ systems, in particular the eye, heart, kidneys, gut, and skeleton.
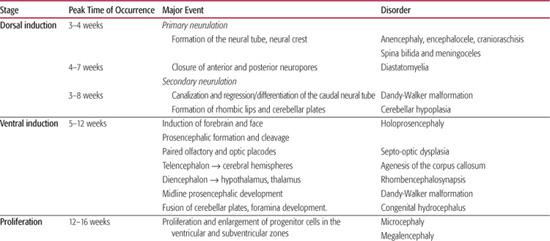
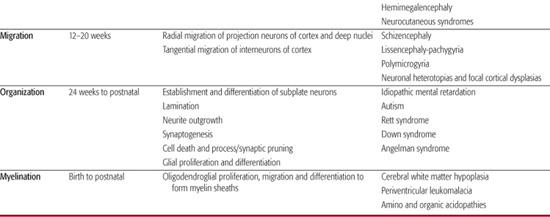
The neuraxis develops following the fate decision of several early embryonic cells to become neural progenitors and by the second week of embryogenesis the three primary layers of ectoderm, mesoderm, and endoderm are formed. It subsequently proceeds through the dorsal and ventral induction to form the neural tube, lower spinal cord and eventually the prosencephalon (forebrain), mesencephalon (midbrain), and rhombencephalon (hindbrain). This is followed by massive cellular proliferation of both neuronal and glial precursors and later differentiation into specific neuronal and glial types. Cortical development occurs with migration of neuronal and glial precursors away from the ventricular and subventricular zone towards the pial surface to form the neocortex and major commissures such as the corpus callosum. This is followed by cortical organization that includes alignment, orientation, and layering of cortical neurons together with synaptogenesis. From the end of the second trimester into early childhood late glial differentiation and myelination, together with programmed and experience-dependent synaptic formation and pruning predominate.
The range of known malformations is almost as complex as the series of developmental events that lead to the formation of the brain. They are perhaps best understood as the effects of insults—regardless of type—at key points in the development of the brain. Although much emphasis has been placed on genetic etiologies—cytogenetic abnormalities, single gene disorders, and polygenic syndromes—environmental insults at key points in the developmental program may result in similar malformations with common clinical findings in the infant or child. As the developing brain, particularly during the first two trimesters, is not capable of generating the common glial response to injury, the etiological cause is frequently difficult to discern. For example early vascular or infectious events may cause abnormalities of cortical migration such as polymicrogyria indistinguishable from a primary genetic causation. An overview of the key developmental events in brain and spinal cord formation, together with examples of corresponding disorders related to developmental changes at these steps is given in Table 548-1.
The clinical sequelae of these malformations are broad and often location specific, but some general themes emerge. Disorders of embryonic CNS development (neurulation, prosencephalic development, and neuronal proliferation) are frequently associated with morphological findings. These include abnormal head size and shape; facial dysmorphology (in particular midline abnormalities); facial clefting; and anomalies of optic globe size, shape, and position. Intra-axial anomalies frequently lead to major variations in ventricular size and structure and consequently these lesions are diagnosed on routine obstetric fetal anomaly scans. Spinal closure abnormalities may be associated with external stigmata such as fistulae, dimples, nevi or ectopic hair, or fat. In addition fetal lower limb positioning may be abnormal with talipes and fixed flexion of the hips of extension of knees detectable prenatally. In addition to fetal miscarriage and death, other fetal manifestations of early CNS malformations include polyhydramnios, decreased fetal movement, and preterm labor. Malformations related to abnormalities in later fetal stages of CNS formation may present postnatally, often in late infancy or childhood, frequently without evident dysmorphic features or neonatal manifestation. Early symptoms may include hypotonia and motor delay, as well as feeding difficulties. Later symptoms usually encompass speech and language delay and variable degrees of cognitive impairment including mental retardation. Epilepsy is particularly associated with disorders of migration and cortical organization but may not present until late childhood or adolescence. This chapter covers developmental disorders up to and including disorders of migration. Conditions associated with aberrant organization, synaptogenesis, pruning, and myelination are covered elsewhere in detail.
Table 548-1. Major Events in Human Brain and Spinal Cord Formation and Corresponding Disorders in Development
ABNORMALITIES OF CRANIAL DEVELOPMENT
 ANOMALIES OF PROSENCEPHALIC DEVELOPMENT
ANOMALIES OF PROSENCEPHALIC DEVELOPMENT
The developing forebrain and face is contingent on the ventral induction by the prechordal mesoderm at the rostral end of the neural tube. The sonic hedgehog signaling pathway is critical to the development of the prosencephalon, with sonic hedgehog protein (Shh) secreted from the prechordal mesoderm to activate the Patch receptor and downstream genes.
The major events are formation of the prosencephalon at the end of the first month; cleavage in three planes to give the basic structure of paired cerebral hemispheres, basal ganglia, and ventricles; separation from the midbrain; and development of paired optic and olfactory structures. These events are followed by midline prosencephalic development with thickening of the commissural, chiasmatic, and hypothalamic plates necessary for formation of the corpus callosum, septum pellucidum, optic nerve chiasm, and the hypothalamus. The corpus callosum forms as developing cortical axons cross the midline under the influence of chemoattractants and repellents.
 HOLOPROSENCEPHALY
HOLOPROSENCEPHALY
This group of disorders is the result of different degrees of failure of prosencephalic cleavage and distinguished by the severity of failed cleavage of the cerebral hemispheres and deep nuclear structures. The most severe form is alobar holoprosencephaly where there is a single sphere cerebral structure and monoventricle with fusion of the thalami and deep nuclei together with absence of the corpus callosum and olfactory bulbs. Neuronal migration and cortical organization is severely disordered. In semi-lobar holoprosencephaly there is failure of separation of the anterior cerebral cortex and absence of the anterior corpus callosum. The least severe forms are lobar holoprosencephaly, in which the cerebral hemispheres are fully separated and the deep nuclei partially separated, and the middle interhemispheric variant, in which only the posterior frontal and parietal regions fail to separate. Hydrocephalus is present in most infants with alobar holoprosencephaly due to fusion of the thalami and impaired drainage of cerebrospinal fluid (CSF) through the aqueduct. Infants with semilobar and lobar forms are usually microcephalic. Facial anomalies are usual and range from severe cyclopia with proboscis through to ocular hypotelorism with a flat single nostril nose (cebocephaly) to mild hypotelorism with or without cleft palate. Malformations of other organ systems are present in 75% of patients.
Alobar holoprosencephaly occurs in 1/10,000 live births but is 100 times more common in conceptuses examined after miscarriage or abortion. Causes include both genetic and cytogenetic etiologies as well as teratogens. Over two thirds are related to full or partial chromosomal aneuploidies, in particular trisomy 13. A list of known etiologic factors is given in Table 548-2.
The clinical features relate to the severity of failure of cleavage of the cerebral hemispheres, basal ganglia, and abnormal hypothalamic function. Most patients with alobar holoprosencephaly do not survive infancy. Children with less severe forms do have prolonged survival. Seizures are common as is profound visuomotor and cognitive impairment. The failure of basal ganglia and thalamic cleavage leads to dystonia and motor impairment, as well as early apnea. Hypothalamic dysfunction leads to often life-threatening endocrinopathies such as diabetes insipidus, as well as poikilothermia. Management is primarily supportive. Although shunting is performed, the development of hydrocephalus is often a sign of unrecognized severe failure of cleavage.
 AGENESIS OF THE CORPUS CALLOSUM AND ABSENT SEPTUM PELLUCIDUM
AGENESIS OF THE CORPUS CALLOSUM AND ABSENT SEPTUM PELLUCIDUM
This group of disorders is characterized by varying degrees of failure of midline prosencephalic development. Agenesis of the corpus callosum (ACC) may be complete or partial and is associated with deformation of the lateral ventricles to give a parallel ventricular arrangement known as colpocephaly. This is usually accompanied by longitudinal fibers coursing along the medial aspect of the hemispheres known as Probst bundles. Partial ACC usually comprises absence of the posterior callosum. Agenesis of the corpus callosum (ACC) may be isolated or in up to 40% to 50% of cases associated with other cerebral abnormalities, specifically cerebellar malformation and cortical migration anomalies. Agenesis of the corpus callosum is one of the most common anomalies occurring in up to 7 in 1000 live births and 3% of children with developmental delay. It is increasingly diagnosed in fetal life. Agenesis of the corpus callosum may be completely asymptomatic or have symptoms only detected on very refined testing of interhemispheric transfer. In association with other anomalies however there is a strong risk of cognitive and neuromotor impairment. Agenesis of the corpus callosum is associated with well over 65 known genetic syndromes and the presence of extra-axial abnormalities or cytogenetic abnormalities is a risk factor for impaired neurologic development (Table 548-3). A notable condition is Aicardi syndrome, an X-linked dominant condition affecting females associated with neuronal heterotopias, chorioretinal lacunes, early onset infantile spasms and subsequent refractory epilepsy and profound developmental delay.
Table 548-2. Etiologic Factors in Holoprosencephaly



