Conjugated Hyperbilirubinemia
EPIDEMIOLOGY
Historical estimates place the incidence of cholestasis among all neonates at 1 in 2500 live births.1 A recent retrospective data review found that among all infants cared for in a large hospital system in the United States who had at least 1 measured conjugated bilirubin level, 3 in 1000 had a peak conjugated (direct) bilirubin greater than 2.0 mg/dL2; however, in the majority of these patients a specific hepatobiliary disorder was not identified, and the peak bilirubin was relatively low (<5 mg/dL). The incidence of cholestasis in the neonatal intensive care unit (NICU) is significantly higher. In 1 series, cholestasis (defined as direct bilirubin > 1.0 mg/dL when total bilirubin is 5 mg or less or direct bilirubin > 20% of total bilirubin when the total bilirubin is above 5 mg/dL) was identified in 2% of all NICU patients.3 Risk factors that are common in this population include prematurity, low birth weight, sepsis, shock, surgery, and parenteral nutrition.4 Thus, although cholestatic liver disease is rare in general outpatient pediatrics, the neonatologist must be familiar with the initial triage and diagnostic evaluation, as well as supportive care, of the cholestatic infant.
PATHOPHYSIOLOGY
Cholestasis refers to any impairment in the hepatic excretion of bile. Bile is an aqueous solution of bile acids, cholesterol, conjugated bilirubin, proteins, phospholipids, toxins and xenobiotic substances, and electrolytes. Biochemically, an increase in serum conjugated bilirubin is the most commonly identified marker of cholestasis. The usual rule of thumb is that a direct-reacting fraction of bilirubin greater than 2 mg/dL or greater than 15% of total bilirubin should be regarded as evidence of cholestasis. However, it is helpful to remember that direct and conjugated bilirubin are not identical. Unconjugated bilirubin is the product of heme breakdown, which in the liver is conjugated with glucuronic acid prior to secretion into bile. The traditional assay relies on the fact that conjugated bilirubin (the “direct-reacting fraction”) reacts much more readily with a diazo reagent than does unconjugated bilirubin (the “indirect-reacting fraction”), which is measured after the addition of an accelerating agent. Unconjugated bilirubin does participate, albeit more slowly, in the unaccelerated reaction, and when the concentration of unconjugated bilirubin is high, the direct bilirubin measurement will be spuriously elevated.5,6 Laboratories that report conjugated and unconjugated fractions as such, rather than as direct and indirect fractions, are typically using a newer spectrophotometric method (Kodac BuBc), and some laboratories report measurements for both direct and conjugated bilirubin. The direct bilirubin measurement (ironically, an indirect estimate of the conjugated bilirubin concentration) is generally a higher number than the conjugated bilirubin measurement, and it also includes measuring the delta bilirubin or the bilirubin bound to albumin.
Physiology of Normal Bile Production and Excretion
Some understanding of bile acid physiology is necessary for an adequate description of cholestatic pathophysiology. The biochemistry and transport of bile acids is complex, and this discussion is restricted to the clinically salient points. Figure 39-1 provides a cartoon representation of the basic machinery of bile excretion.
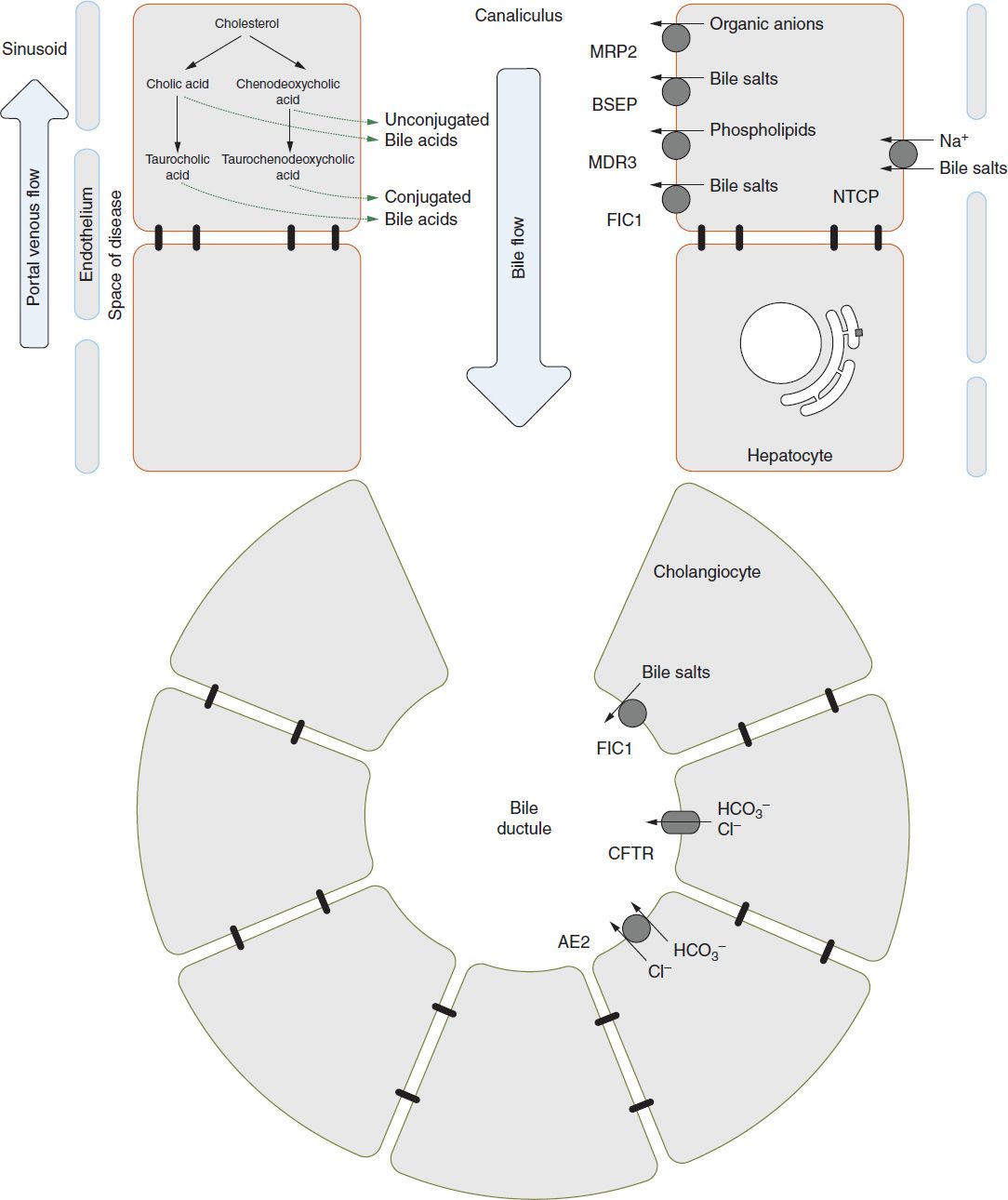
FIGURE 39-1 Basic anatomy of the hepatocytes and cholangiocytes as it relates to bile formation and enterohepatic circulation. The specific transporters are discussed in the text. BSEP, bile salt excretory protein; FIC1, familial intrahepatic cholestasis 1.
Bile Acid Synthesis and Enterohepatic and Cholehepatic Circulation
Bile acids are synthesized from cholesterol within hepatocytes. Cholic acid and chenodeoxycholic acid are the bile acids synthesized by the liver and are referred to as “primary” bile acids. After conjugation with glycine or taurine, they are secreted with other normal bile constituents into the duodenum. Bacterial action within the small bowel converts cholic acid to deoxycholic acid and chenodeoxycholic acid to lithocholic acid, the “secondary” bile acids. The majority of intestinal bile acid is reabsorbed through an active transport mechanism in the ileum, transported to the liver via the portal vein, and again taken up by hepatocytes.7 This cycle is referred to as the enterohepatic circulation.
Functions of Bile Acids
Bile acids perform myriad physiologic functions. Within the hepatocyte, they participate in the regulation of multiple genes by means of the farnesoid X receptor, a nuclear receptor also known as the bile acid receptor.8 In bile, they form the major osmotic force driving the secretion of water, increasing bile volume and flow rate (so-called bile-acid-dependent flow), and thus an adequate pool of bile acids is required for normal bile excretion.9 Fecal loss of bile acids provides a major route for the elimination of cholesterol in humans, and bile acids solubilize unmodified cholesterol in bile as well. Bile acids function within the intestine to promote diffusion and absorption of fatty acids and lipid-soluble vitamins (A, D, E, K). They have direct antimicrobial activity and appear to regulate other antimicrobial activity within the small intestine mediated by FXR receptors. In the colon, bile acids stimulate propulsive motility as well as modulate water and electrolyte transport. There is also intriguing evidence that bile acids act as endocrine factors via G protein-coupled receptors to modulate adipocyte thermogenesis and hepatic blood flow.7
Physiologic Cholestasis of the Neonate
The normal physiology of the neonatal liver differs from that of older children and adults in clinically important ways, particularly with regard to the processing and excretion of bile. Evidence collected from human and animal studies has led to the concept of a “physiologic cholestasis” of infancy.10 Compared to older infants and adults, the neonate possesses a decreased total-body pool of bile acid, and this pool is relatively concentrated in the hepatic and circulating compartments rather than within the bowel lumen. After birth, the rates of bile acid synthesis and uptake by hepatocytes increase rapidly, as does the intraluminal concentration of bile acids within the bowel. The concentrations of bile acids in serum decline more slowly to normal adult levels over the first 6–12 months of life.11–13 These findings are thought to reflect immaturity in the uptake and synthesis of bile acids by hepatocytes and in the transport of bile acids across the ileal epithelium.
Consequences of Cholestasis
The result of cholestasis is retention of bile acids and bile products within the hepatocytes and liver. As the level of these products builds, damage to the hepatocytes results.
Hepatic Injury and Biliary Cirrhosis
As the intrahepatic level of bile salt and bile contents increases, there is activation of Kupffer cells and other cells to produce fibrosis.14,15 Over time with chronic cholestasis, the typical periportal fibrosis accompanied by bile stasis and loss of bile ducts is characteristic of biliary cirrhosis. The fibrosis gradually increases within and between portal tracts.
Malabsorption and Malnutrition
Bile acids act as a detergent within the bowel lumen, solubilizing lipids and allowing their absorption by enterocytes. Because of the decreased concentration of bile acids in the bowel, some degree of steatorrhea is normal in neonates and is more pronounced in infants fed cow’s milk–based formula because it lacks the lipase activity found in human milk.16 Pathologic cholestasis exacerbates this fat malabsorption and predisposes the infant to fat-calorie malnutrition as well as deficiency in the fat-soluble vitamins (A, D, E, and K).17 One patient series found a high prevalence of fat-soluble vitamin deficiency among children waiting for a liver transplant, with a majority of patients having a deficiency in at least 1 of the vitamins A, D, E, and K.18
In addition to malabsorption and secondary deficiencies in specific micronutrients, children with chronic liver disease are likely to have higher resting energy expenditure than their healthy age cohort.19,20 Thus, supplemental enteral feeding is sometimes required in chronic liver disease to maintain normal growth.
Pruritus
Pruritus in chronic cholestasis presumably results from the activity of an unknown “pruritogen,” which is normally excreted in bile and that at elevated levels can stimulate either peripheral or central itch pathways.21 Although there do not appear to be any published data regarding pruritus in neonates, it is a widespread and sometimes-debilitating problem among older patients with cholestatic liver disease.22
DIFFERENTIAL DIAGNOSIS
Because of the unique susceptibility of the neonatal liver, cholestasis is often a nonspecific response to systemic stress, resulting in a self-limited “idiopathic neonatal hepatitis.” However, it may also be the presenting sign of serious infectious or metabolic disease or of biliary tract anomalies that require surgical attention. To make matters more complicated, the neonatologist and gastroenterologist are often faced with an infant whose history includes prematurity, septic or ischemic insults, and recent or ongoing reliance on parenteral nutrition, all of which may contribute to cholestasis. In such cases, the decision regarding how extensively, and when, to search for other possible causes of cholestasis is a difficult one. Although it is almost impossible to enumerate all disorders that can result in neonatal cholestasis, a neonatologist is likely to find it helpful to know those entities that are common, require urgent diagnosis and treatment, or respond to specific therapy (Table 39-1).
Table 39-1 A Somewhat Comprehensive List of Diagnoses That May Lead to Cholestasis in the Neonate


As Figure 39-2 illustrates, the diagnostic categories of neonatal cholestasis have been refined over the past 4 decades. Research into the biochemistry and genetics of bile formation, often enabled by individual patients or families with inherited cholestatic disorders, has broken the previous majority diagnosis of “idiopathic neonatal hepatitis” into multiple specific genetic and metabolic entities.23
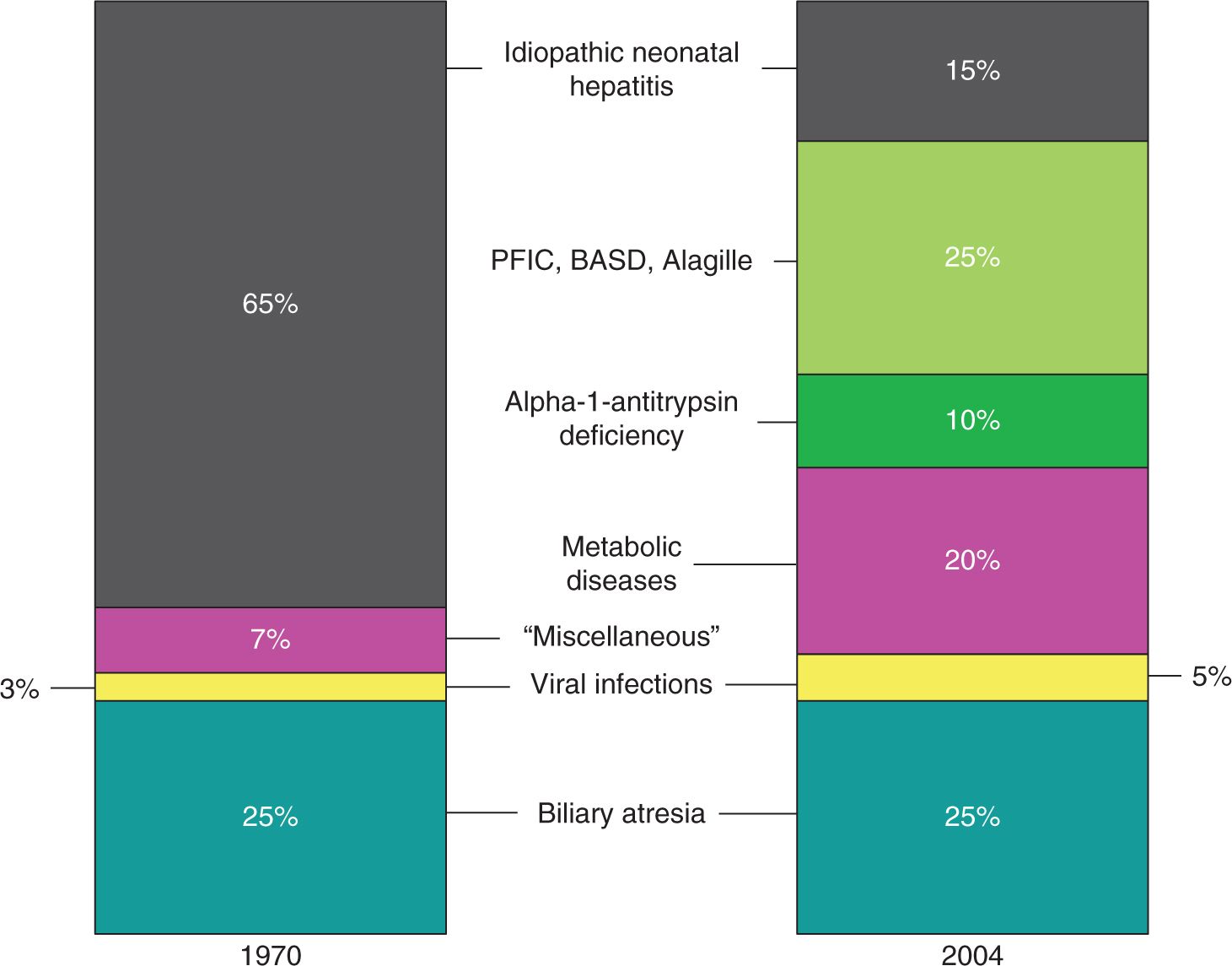
FIGURE 39-2 Major diagnoses resulting in neonatal cholestasis as of 1970 and 2004. Observe that the proportions of biliary atresia and viral hepatitis have remained constant. The previously large group of infants who were labeled with having “idiopathic neonatal cholestasis” has been broken down into multiple genetic disorders. The majority of these infants with chronic cholestasis can now be successfully diagnosed given sufficient time and resources. AAT, α1-antitrypsin. (Adapted with permission from Balistreri WF, Bezerra JA. Whatever happened to “neonatal hepatitis”? Clin Liver Dis. 2006;10(1):27–53.)
Obstructive Disorders
Extrahepatic
Biliary obstructive disorders, either intrahepatic or extrahepatic, make up the largest proportion (50%) of cases of conjugated hyperbilirubinemia in infants for which a single cause can be identified.24 Biliary atresia is the most common diagnosis in this group, which includes other “anatomic” problems of the bile ducts, such as choledochal cysts, choledochoceles, inspissated bile syndrome, strictures, and cholelithiasis. Both intrahepatic and extrahepatic biliary obstruction may occur as a result of compression of bile ducts by an extrinsic mass, for example, in hepatoblastoma.
Intrahepatic
There are a number of possible intrahepatic causes. Alagille syndrome due to jagged 1 protein or NOTCH receptor mutations may have significant cholestasis with high cholesterol and γ-glutamyl transferase (GGT) and with paucity of intralobular ducts on liver biopsy.25 There are also some infants with a so-called nonsyndromic paucity of bile ducts. There are a number of cholestatic genetic syndromes that result in abnormal bile flow due to canalicular bile transport mechanisms. One group is the progressive familial intrahepatic cholestasis (PFIC) disorders (Figure 39-1). They are autosomal recessive disorders affecting various transporters. PFIC1 or Byler disease is due to an adenosine triphosphate (ATP)–dependent amino phospholipid transporter or FIC1 (familial intrahepatic cholestasis 1). PFIC2 involves bile salt excretory protein (BSEP), which is an ATP-dependent bile acid transport protein.26 MDR3 defects involving an ATP-dependent phospholipid transport protein are found in PFIC3 patients, who are distinguished by having an elevated GGT and elevated triglycerides.27 Benign recurrent intrahepatic cholestasis (BRIC) is a category of cholestasis also caused by defective BSEP or FIC1 protein function but due to different mutations than in PFIC1/2 and having a milder clinical course.28 Other disorders of intrahepatic cholestasis include Dubin-Johnson syndrome and Rotor syndrome.29 Caroli disease and congenital hepatic fibrosis with portal hypertension occur in patients with autosomal recessive polycystic kidney disease (ARPKD)30 as a result of malformation of the ductal plate and embryologic stage in the formation of the liver.31,32 Mutations in CFTR affect chloride and bicarbonate transport and may lead to inspissated bile and the cholangiopathy associated with cystic fibrosis.33,34 Neonatal sclerosing cholangitis is another rare cause of intrahepatic obstruction.35
Hepatocellular Disorders
The other major category of cholestasis is related to hepatocellular injury and dysfunction. Although it is reasonable to categorize some disease as either hepatocellular or obstructive (eg, the PFICs), we have chosen to label them obstructive because they are due to a specific defect in bile transport rather than generalized hepatocellular dysfunction.
Infections
Viral hepatitis represents only 5% of neonatal cholestasis but is important to consider, as are other infectious hepatitides, since specific pharmacologic therapy may be necessary. A large list of viruses may cause neonatal cholestasis or hepatitis: cytomegalovirus (CMV), herpes simplex virus (HSV), human herpesvirus 6 (HHV-6), parvovirus B19, coxsackie virus, echovirus, and the classic hepatitis viruses A, B, C, and E.24, 36, 37 Nearly any bacterial, protozoal, or fungal infection severe enough to provoke a systemic inflammatory response can cause cholestasis, but several in particular should be considered: occult urinary tract infection, gram-negative sepsis, tuberculosis, syphilis, and listeria; fungemia due to central line contamination; and toxoplasmosis.
Metabolic Disorders
Metabolic diseases as a group cause some 20% of neonatal cholestasis, but a large number of specific diagnoses fall into this category, each of which is rare. Thus, if a preliminary biochemical screening produces signs of an inborn error of metabolism, it is essential to involve a medical geneticist early in the workup. Some of the more common disorders are discussed here.
α1-Antitrypsin (AAT) deficiency of the ZZ and SZ phenotypes predisposes to progressive liver injury and cholestasis, which can begin in infancy. AAT deficiency by itself is among the more common causes of neonatal cholestasis (20%).38 Other common inborn errors of metabolism that can cause cholestasis include galactosemia, hereditary fructose intolerance, and tyrosinemia.39 Galactosemia can present in the first days of life with lethargy, hepatic dysfunction, and lactic acidosis. Galactosuria can be identified by a positive test for urinary reducing substances. Hereditary fructose intolerance produces hypoglycemia, gastrointestinal symptoms, and liver and renal tubular dysfunction but should not present until the introduction of fructose-containing foods, typically after weaning. Untreated tyrosinemia results in severe liver injury that can cause acute liver failure or rapidly progressive cirrhosis with cholestasis and portal hypertension within the first year. Older children may have recurrent bouts of severe pain, cirrhosis, and hepatocellular carcinoma. Because outcomes are significantly improved with medical treatment in all of these disorders, it is important to make the diagnosis promptly.
The remaining metabolic disorders that can lead to cholestasis are extremely numerous but can be divided into several broad categories: mitochondrial respiratory chain disorders; fatty acid oxidation disorders; peroxisomal disorders; disorders of amino acid metabolism, including urea cycle defects; bile acid synthetic defects; congenital disorders of glycosylation; and glycogen storage diseases (see Table 39-1).
Endocrine Disorders
Panhypopituitarism, isolated cortisol deficiency, and perhaps hypothyroidism can all cause neonatal cholestasis.40,41 Since rapid medical treatment is beneficial, an endocrine screening is recommended early in the evaluation.
Toxic Causes
Toxic hepatic injury can be the result of maternal use of medications or illicit drugs, as well as an iatrogenic illness caused by medications given to the infant after birth. Perhaps the most important of the toxic etiologies is intestinal failure-associated liver disease (IFALD; total parenteral nutrition [TPN] cholestasis).42 Infants with intestinal failure develop cholestasis and cirrhosis, probably due the combined effects of toxic elements in TPN (eg, lipid emulsion) and recurrent bacterial infection or translocation. IFALD is a major cause of end-stage liver disease requiring transplant, but new therapeutic approaches discussed in the management section can help to normalize liver function and may reduce the need for transplantation in the future. Cholestatic liver injury due to medications is often idiosyncratic and difficult to predict, but there has been a suggestion that fluconazole may predispose neonates to cholestasis.43,44 Other agents more commonly associated with cholestatic liver injury in older patients should also be considered, such as amoxicillin-clavulanate, macrolides, and nonsteroidal anti-inflammatory drugs (NSAIDs).45
Prenatal exposure to certain agents, such as carbemazepine46–48 and methamphetamine,49 has also been associated with neonatal cholestasis.
Immunologic Causes
Immune-mediated injury to the hepatocytes is responsible for some causes of neonatal cholestasis. Examples include neonatal lupus erythematosus50–52 and neonatal hemochromatosis (neonatal iron storage disorder),53 caused by maternal antibodies, and hemophagocytic lymphohistiocytosis (HLH).54
Genetic Disorders
There are a number of genetic disorders that may present with cholestasis. Trisomy 18 and 21 are examples with dysmorphic features. Other examples are the ARC syndrome (arthrogryposis-renal dysfunction-cholestasis syndrome) and the Kabuki syndrome (associated with cleft palate).
Hypoxia or Hypoperfusion Injury
Infants who suffer hypoxic or hypoperfusion injury due to congenital or acquired cardiopulmonary disease,55,56 shock, or hepatovenous obstruction (Budd-Chiari syndrome) will often present with jaundice. Frequently, these infants will have other evidence of end-organ injury, such as renal disease.
Other Causes
There are a few other disorders found in specific ethnic groups, such as Aagenaes syndrome (lymphedema with cholestasis syndrome)57,58 and North American Indian childhood cirrhosis.59
Figure 39-3 presents some of the more important diagnoses leading to neonatal cholestasis according to anatomic location to illustrate the scheme used for organizing these disorders.
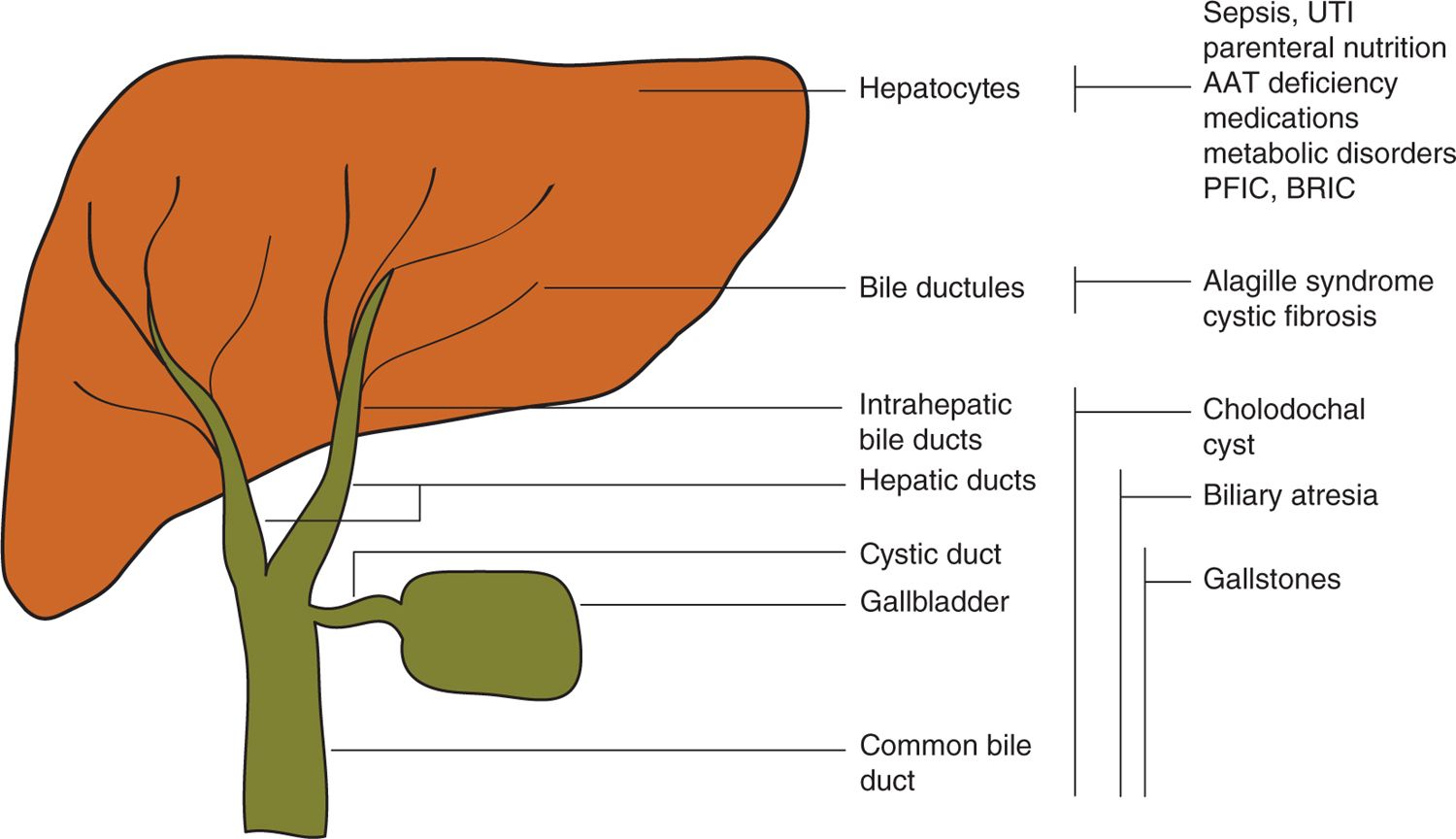
FIGURE 39-3 Common disorders, arranged according to anatomic site, leading to neonatal cholestasis. AAT, α1-antitrypsin; BRIC, benign recurrent intrahepatic cholestasis; PFIC, progressive familial intrahepatic cholestasis; UTI, urinary tract infection.
DIAGNOSTIC TESTS
Initial Evaluation of the Cholestatic Infant
In the general pediatric setting, cholestasis is usually first identified by the presence of visible jaundice. Although most otherwise-healthy term infants with jaundice have self-limited unconjugated hyperbilirubinemia of benign origin, it is important to identify those few with conjugated hyperbilirubinemia. A consensus guideline from the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition (NASPGHAN) recommends obtaining fractionated bilirubin levels in infants who remain visibly jaundiced past 2 weeks of age.1 This is partly in response to the typical schedule of well-baby visits in the United States, according to which an infant who is healthy at 2 weeks of age may not normally be seen again by the pediatrician until 2 months of age. Two months is an unacceptably long delay for an infant who is subsequently found to have cholestatic jaundice, particularly given that the prevailing wisdom is that outcomes for surgical management of biliary atresia become significantly worse between 2 and 3 months of age.60,61 Newly published evidence suggests that early elevation of direct bilirubin (within 48 hours of birth) may identify infants with biliary atresia.62 However, it remains to be seen whether biliary atresia can reliably be excluded on the basis of a normal fractionated bilirubin level at such a young age. In the NICU, cholestasis is frequently identified by laboratory screening in the absence of visible jaundice. Occasionally, evidence of cholestasis is specifically sought because there is suspicion of a disease that may entail cholestasis, for example, if there are morphologic signs of Alagille syndrome or a positive screening test for cystic fibrosis.
After confirming the presence of cholestasis by conjugated and unconjugated (or direct and indirect) bilirubin levels, the clinician is faced with 2 immediate questions. The first relates to the degree of illness. Since cholestasis may be a sign of severe infection, shock, or metabolic disease, it is important to determine if the cholestatic infant is in need of immediate specific treatment. Usually, this triage can be made on the basis of information already in hand, such as the clinical history, vital signs, and overall clinical assessment. Second, an initial assessment of liver function is important at this stage as well since an infant with impaired function is in need of more urgent diagnostic evaluation and possible listing for liver transplantation. Thus, it is important to obtain or review measures of blood glucose, ammonia, albumin, and coagulation times.
In an infant who is generally well, with preserved hepatic function, the clinician can then evaluate in a stepwise fashion. Table 39-2 presents a reference list of tests that the physician will need to consider ordering, arranged somewhat arbitrarily into stages according to when they might typically be appropriate. Of course, the severity of illness, the age of the infant, and the presence of diagnostic clues in the history and physical will influence the choice of tests in particular patients.
Table 39-2 Tests Often Used in the Evaluation of Neonatal Cholestasis
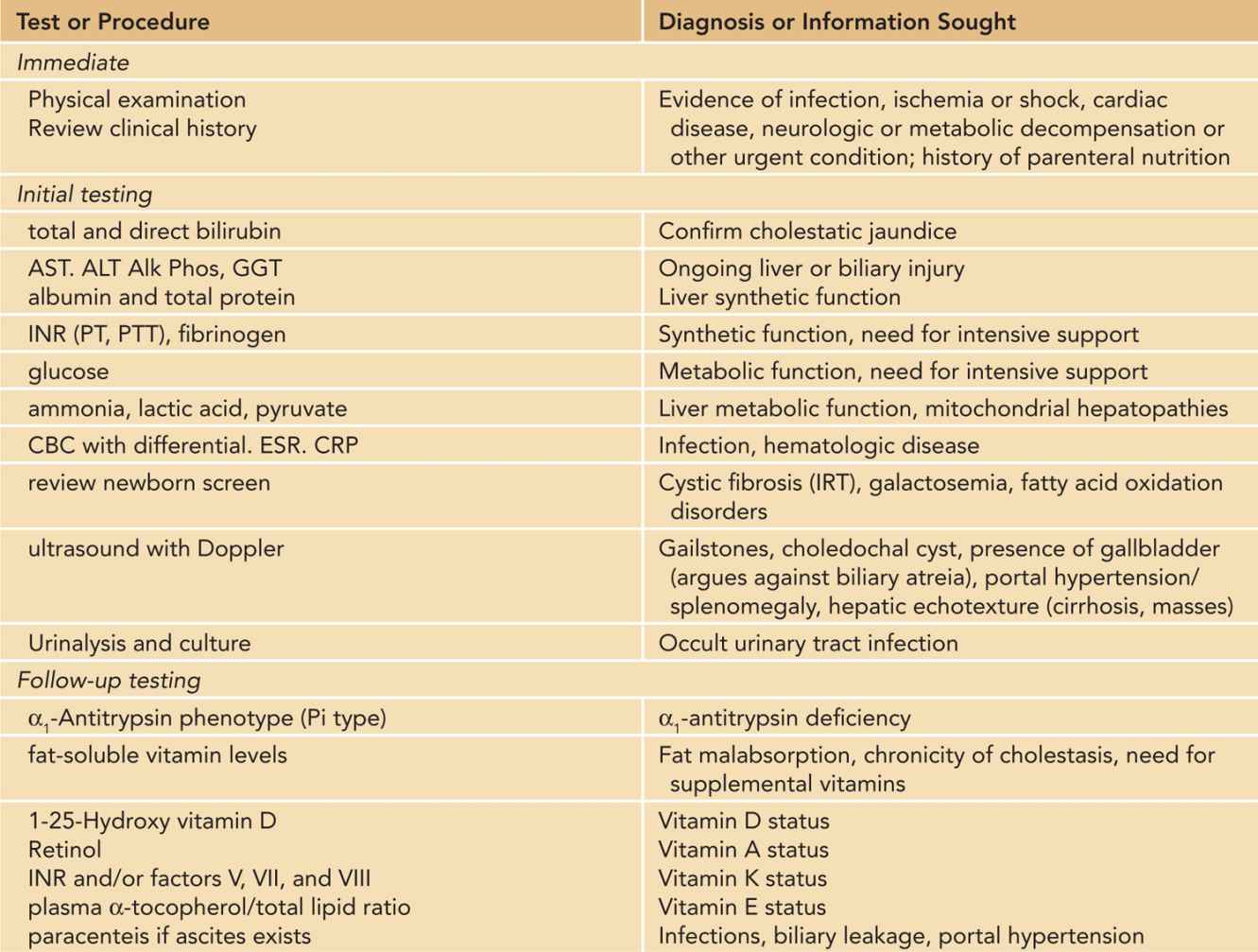
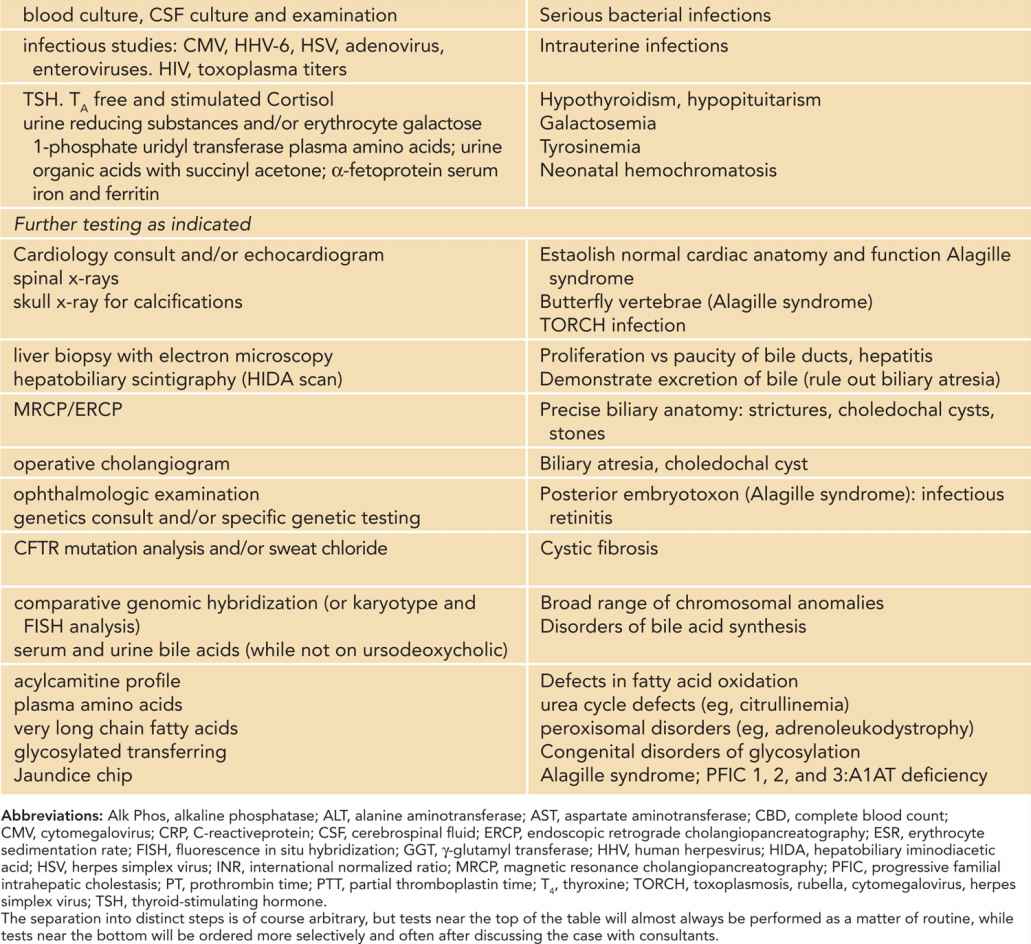
The initial step in the diagnostic workup usually will include a complete biochemical evaluation for liver injury (AST [aspartate aminotransferase], ALT [alanine aminotransferase], alkaline phosphatase, GGT), as well as selected specific diagnostic tests, such as ultrasound of the liver and gallbladder, and urinalysis and culture. Depending on the degree of suspicion, other serious bacterial infections will need to be ruled out in the initial phase.
History
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


