Congenital Heart Disease
Julien I. E. Hoffman
INCIDENCE
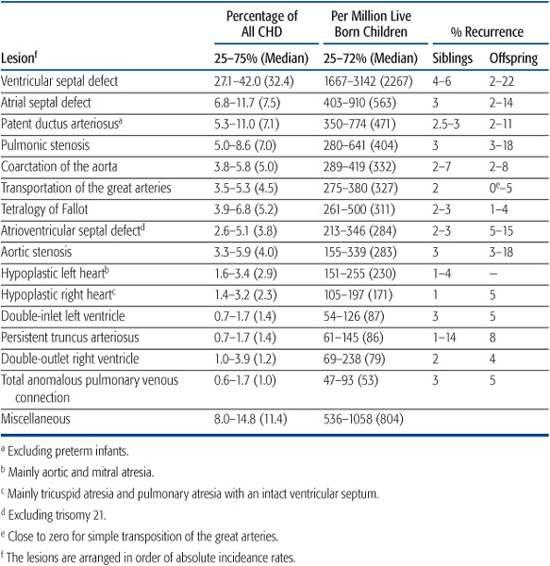
Congenital heart diseases occur in at least 10 per 1000 live-born children; the incidence is much higher in stillborn infants and in spontaneous abortuses. This figure excludes bicuspid aortic valves, patent ductus arteriosus in premature infants, and tiny muscular ventricular septal defects with respective incidences of 10 to 20, 4 to 5, and 30 to 40 per 1000 live-born children. The distributions of various common types of congenital heart diseases at birth are given in Table 484-1.
Table 484-1. Relative and Absolute Incidence of Major Congenital Heart Lesions and Their Recurrence Rates
ETIOLOGY
Congenital heart diseases result from interaction between genetic and environmental factors.
Genetic Factors Single classic mendelian mutant genes account for 3% of congenital heart diseases; 5% are caused by gross chromosomal aberrations, 3% by known environmental factors (eg, rubella, fetal alcohol syndrome), and the rest by multifactorial gene effects or single gene effects modulated by random events. The genetics of congenital heart disease are discussed in detail in Chapter 481.
Environmental Factors Women taking lithium salts during pregnancy may have children with congenital heart diseases, with a high incidence of mitral and tricuspid valve lesions, especially Ebstein syndrome. Diabetic women or those taking progesterone in pregnancy have an increased risk of having children with congenital heart diseases. About half the children of alcoholic mothers have congenital heart diseases (usually left-to-right shunts). Retinoic acid used to treat acne may cause several types of congenital heart lesions.
Viral Lesions Rubella embryopathy is often associated with peripheral pulmonic stenosis, patent ductus arteriosus, and valvar pulmonic stenosis. Other viruses, notably coxsackieviruses, have been thought to cause congenital heart diseases because of an increased frequency of rising serum titers to this virus in mothers whose infants have congenital heart disease. Mumps virus is responsible for endocardial fibroelastosis.
COUNSELING FAMILIES
The risk of occurrence of cardiac lesions in future children concerns parents. Chromosomal abnormalities have risks of recurrence that vary with the specific chromosomal change involved. Other forms of inheritance produce a much lower risk of recurrence (Table 484-1). Furthermore, if 2 first-degree relatives have congenital heart disease, then the risk of heart disease in the next infant is about 3 times as high as the figures just cited. The risk of transmission of congenital heart disease to children if the parent, especially the mother, has congenital heart disease averages about 5% to 10%. If another child has congenital heart disease, it is most often similar in type (concordant) to that in the parent or sibling.1-9
When a child is found to have congenital heart disease, the parents frequently have severe guilt feelings and are almost always worried about the risk of occurrence of congenital heart disease in future children. These issues should be discussed openly with the parents, who are often reticent about mentioning them. An explanation of what is known of the causes of congenital heart diseases and reassurance that the parents did not cause it by acts of omission or commission are arguments that can be used to help allay guilt feelings. This approach must be correlated with all other aspects of giving continued support to parents with chronically ill children.
LEFT-TO-RIGHT SHUNTS
A shunt from systemic to pulmonary circulation through an abnormal communication, termed a left-to-right shunt, lets oxygenated blood recirculate through the lungs without entering the peripheral arterial circulation. This shunt is wasted flow that adds to cardiac work without improving delivery of oxygenated blood. A left-to-right shunt may be present alone or associated with right-to-left shunting (bidirectional shunting) or obstructive lesions.
Left-to-right shunts are classified anatomically by the level at which the systemic and pulmonary circulations communicate.
With defects that produce left-to-right shunts after birth, fetal somatic development is unaltered, and blood flow to the fetal organs and placenta is probably normal. However, alterations of flow patterns in the fetal heart and great vessels may affect their development. When some left ventricular output is shunted away from the ascending aorta, as may occur in endocardial cushion defects, a large ventricular septal defect, or double-outlet right ventricle, particularly with aortic or subaortic obstruction, then decreased aortic isthmus flow may cause hypoplasia or even interruption of the aortic isthmus. Altered streaming patterns may change the composition of blood leaving the heart. Thus, with an endocardial cushion defect or large ventricular septal defect, the oxygen tension of blood leaving the right ventricle and perfusing the lungs may be higher than that in the normal fetus. This higher oxygen tension may alter the development of pulmonary resistance vessels, thereby affecting postnatal clinical features.
Defects associated with left-to-right shunts are shown in Table 483-4. Four major interrelated factors control the amount of left-to-right shunting postnatally: the size of and therefore the resistance to flow offered by the communication, the difference in pressures between the chambers or vessels, the relative distensibilities of the 2 ventricles, and the total outflow resistances (including peripheral resistances) of the chambers or vessels.
If the communication at any level is small, it offers a high resistance to flow through it so that the left-to-right shunt will be small, no matter what the pressures or resistances are. The latter 3 factors come into play only with medium-sized or big communications. The physiology underlying these factors is reviewed in Chapter 483 and in additional text on the DVD. The evaluation and management of specific lesions associated with left-to-right shunts are discussed below. 
A clinically apparent patent ductus arteriosus occurs in 30% to 40% of premature infants with birth weights under 1750 g, and about 80% under 1000 g birth weight. The mechanisms responsible for continued patency are related to the inability of the ductus arteriosus in immature infants to respond normally to an increased oxygen tension and to changes in prostaglandin concentrations. (see Chapter 55). The incidence of persistent patency of the ductus arteriosus in full-term infants born at high altitude is significantly higher than in those born at sea level, probably because of the lower atmospheric oxygen tension. Persistent patency of the ductus arteriosus in full-term and occasional preterm infants at lower altitudes is generally related to a structural abnormality of the ductus arteriosus. 
 CLINICAL MANIFESTATIONS IN MATURE INFANTS
CLINICAL MANIFESTATIONS IN MATURE INFANTS
The diagnosis of patent ductus arteriosus is easier in full-term infants or older children than in immature infants. Because of continuous runoff of blood from the aorta to the pulmonary artery through the ductus arteriosus, the murmur in older infants and children is continuous and has a rumbling, machinery-like or “train in a tunnel” quality, usually with late systolic accentuation of the murmur. It is heard best below the left clavicle. If the ductus arteriosus is small, this may be the only abnormal finding. If it is larger, the increase in left ventricular output is associated with an increase in stroke volume that causes a rapid rise in the aortic pulse pressure as a result of rapid left ventricular ejection and also causes left ventricular hyperactivity. The diastolic runoff through the aortopulmonary communication plus the peripheral vasodilatation from baroreceptor stimulation account for the low diastolic pressure and the collapsing pulse. The increased volume load enlarges the left atrium and ventricle, with roentgenographic evidence of dilatation and electrocardiographic evidence of hypertrophy. Because the ascending aorta receives the increased left ventricular output, it is dilated. On x-ray, the pulmonary vascular markings indicate increased pulmonary blood flow. If there is pulmonary hypertension, there may be signs of right ventricular pressure overload.
The echocardiogram shows the ductus arteriosus and may define its size, as well as assess the volume overload of the left ventricle.
 DIFFERENTIAL DIAGNOSIS
DIFFERENTIAL DIAGNOSIS
In premature infants, particularly those under 1000 g birth weight, there is little chance that clinical findings suggestive of a patent ductus arteriosus are caused by some other congenital heart defect, because patency of an immature ductus arteriosus is so much more frequent than any other form of congenital heart disease. However, in larger premature and full-term infants, sometimes a patent ductus arteriosus cannot be differentiated clinically from aortopulmonary window, truncus arteriosus, ventricular septal defect with aortic regurgitation, or arteriovenous fistula. A major problem may occur when there is severe heart failure with a markedly reduced cardiac output and sympathetic vasoconstriction; the peripheral pulses may not be bounding, the murmur may be soft and not continuous, and the precordium may not be hyperactive. After appropriate therapy for left ventricular failure, the classic physical findings usually reappear.
 OUTCOME
OUTCOME
In the full-term infant with a patent ductus arteriosus, spontaneous closure may occur, but much less commonly than in the premature infant. Medical management, if needed, should be instituted, and at a convenient time surgical closure should be done. Even if there is no heart failure, there are 2 reasons to close a patent ductus arteriosus. If there is marked pulmonary hypertension as a result of a large communication, the danger of the development of pulmonary vascular disease necessitates closure, preferably before 6 to 8 months of age. In the older child with a small patent ductus arteriosus, closure should still be advised in view of the risk of infective endocarditis, even though this risk is very low. Transcatheter closure with a coil is satisfactory if the diameter of the ductus is below 3 mm, but larger ductuses can often be closed by devices such as the Amplatzer ductus occluder. Some ductuses still need surgery, either because they are too large for a catheter-introduced device or, conversely, in a premature infant whose blood vessels are too small to accept the large catheter needed to introduce coils or other devices. Surgery can be done safely by open thoracotomy or by thoracoscopy and may be a much shorter procedure than interventional catheterization.
AORTOPULMONARY FENESTRATION
Aortopulmonary fenestration or window, caused by failure of formation of the base of the spiral septum, generally produces a large aortopulmonary communication just above the semilunar valves. The pulses are typically bounding or collapsing, like those of a large patent ductus arteriosus. However, the murmur more closely resembles that of a high ventricular septal defect in that it is generally not continuous, has a rough, often crescendo-decrescendo character, and is heard maximally along the left sternal border in the third and fourth intercostal spaces. The diagnosis can be made by 2-dimensional echocardiography and confirmed by cardiac catheterization and angiocardiography. Surgical closure during cardiopulmonary bypass is corrective. Occasionally a small opening can be closed by interventional catheterization.
ANOMALOUS ORIGIN OF LEFT CORONARY ARTERY
See below for a detailed discussion.
ANOMALOUS ORIGIN OF PULMONARY ARTERY BRANCH FROM ASCENDING AORTA AND LOBAR SEQUESTRATION
Anomalous origin of a pulmonary artery branch from the ascending aorta has been called hemitruncus arteriosus, but it is not related embryologically to persistent truncus arteriosus because the embryonic truncus arteriosus septates normally in the former defect, with the opposite pulmonary artery arising from a normal pulmonary valve and main pulmonary artery. Most often, the right pulmonary artery arises from the aorta. In lobar sequestration, a portion of the lung, usually a lobe or part of a lobe, gets its arterial blood supply from an abnormal artery arising from the aorta. The involved pulmonary artery does not communicate with the main pulmonary artery. Pulmonary vascular resistance and the resistance offered by the communicating vessel control the flow into the lung or portion of lung. The clinical presentation in children with these lesions also depends on the magnitude of the shunt and will be similar to that found with a patent ductus arteriosus, but congestive heart failure is more common in the anomalous pulmonary artery because it is equivalent to a large patent ductus arteriosus. However, the murmur, often continuous, may be better heard more laterally or even in the back.
One important feature of the anomalous pulmonary artery circulation is that the normally arising pulmonary artery carries the total right ventricular output, which is the total systemic venous return. Normally this flow is distributed between the 2 pulmonary arteries, but with an anomalous pulmonary artery all the systemic venous return passes through about one half of the total number of lung vessels. Pressure in the normally arising pulmonary artery is generally normal; therefore, the risks of subsequent pulmonary vascular disease are similar to those in children with atrial septal defects and a pulmonary blood flow about twice normal. However, with an associated left-to-right shunt, blood flow to the normal lung is more than doubled; thus, there may be pulmonary hypertension and subsequent pulmonary vascular disease. The lung supplied by the abnormally arising vessel is at risk not only from increased flow but also from increased pressure, because this lung is perfused at systemic pressure less the pressure decrease offered by the channel. If the abnormally arising pulmonary artery is adequately developed, implantation into the main pulmonary artery can be done. If a significant portion of lung is involved in lobar sequestration, a lobectomy is indicated. 
 UNILATERAL PULMONARY ARTERY
UNILATERAL PULMONARY ARTERY
Although this is not a lesion with a left-to-right shunt, it resembles an anomalous pulmonary artery from the ascending aorta. The right or the left pulmonary artery may be congenitally absent, either isolated or with other congenital cardiac defects, but most often, the artery arose from a ductus arteriosus that subsequently closed. A unilateral pulmonary artery, either left or right, has the same incidence when these are isolated lesions and when they are associated with most cardiac defects; however, if there is a patent ductus arteriosus, then usually the right pulmonary artery is absent, and in the tetralogy of Fallot, the left artery is almost always missing. The lung on the affected side is hypoplastic and supplied by bronchial arteries if the artery is truly absent, so that the chest roentgenogram shows a small hemithorax, no hilar pulmonary artery, and often a diffuse reticular pattern of bronchial collaterals. Because ventilation can still take place on that side, there is wasted ventilation and usually dyspnea on effort.
The chief importance of the lesion is its tendency to produce pulmonary hypertension and pulmonary vascular disease in all except those with the tetralogy of Fallot. Because there is only one pulmonary artery, it receives the total right ventricular output, just as in the anomalous origin of the pulmonary artery from the ascending aorta. Therefore, even if there are no other lesions, that lung receives twice its normal blood flow; if there are left-to-right shunts in addition, it gets more than this. In infancy, before pulmonary arterial muscle has regressed, this increased flow leads to hypertension and can eventually cause severe pulmonary vascular disease, which has been reported in 18% of patients with no other lesions and 88% of those with cardiac lesions.
Diagnosis is made by angiography or magnetic resonance imaging. Treatment is directed at repairing the associated defects and avoiding anything that might affect the pulmonary vessels of the normal lung (eg, living at high altitude or taking contraceptive pills).
SINUS OF VALSALVA FISTULA
Rupture of a sinus of Valsalva into one of the cardiac chambers is secondary to an aneurysm due to a structural abnormality in the sinus. Most commonly these changes involve the anterior (right coronary) aortic valve sinus, and the ruptured aneurysm subsequently produces a communication from the right coronary sinus into the right ventricle or right atrium. Less commonly, rupture involves the noncoronary or the left coronary sinus; rupture into the left atrium or ventricle is discussed below in Aortic Regurgitation. The aneurysmal dilatation of the sinus that precedes rupture is often associated with a ventricular septal defect, a combination particularly common in persons of Asian descent. Connective-tissue disorders such as Marfan syndrome may also have associated aneurysmal dilatation of the aortic sinuses, but these do not rupture. Small fistulas may occur after infective endocarditis, but more extensive rupture usually occurs after trauma or spontaneously with progressive weakening of the sinus. Acute rupture, although more common in young adults, does occur in children. At the time of rupture, there frequently is an episode of acute chest pain and dyspnea, with sudden onset of a murmur and congestive heart failure; however, a more insidious onset has been described. With rupture into the right ventricle, the physical signs resemble those of a patent ductus arteriosus, with a loud continuous superficial murmur along the left sternal border, but with the addition of an increased right ventricular volume load. With rupture into the right atrium, this lesion will behave like an obligatory shunt, and the features are those of a patent ductus arteriosus and an atrial shunt combined. The diagnosis can be made by 2-dimensional echocardiography, including Doppler and contrast echocardiograph. Accurate differentiation from other lesions may require cardiac catheterization and angiocardiography. Surgical closure of the fistula can be done with cardio-pulmonary bypass.
CONGENITAL CORONARY ARTERIOVENOUS FISTULA
In this lesion, a fistula usually passes from one of the coronary arteries directly into the right ventricle (the most common site) or into the right atrium (either directly or through the coronary sinus). Communications with the left ventricle, left atrium, or pulmonary artery are much less common, except for small coronary-pulmonary fistulas that are of little importance.
The most striking clinical feature is a continuous murmur superficial in character and heard best along the lower left sternal border. The murmur is generally maximal in diastole and has very high-pitched components. Ventricular hyperactivity and a mid-diastolic rumble depend on the magnitude of shunting, which is not usually great enough to cause congestive heart failure. A continuous thrill may be palpable. Occasionally the fistula causes a myocardial steal and ischemic symptoms. The diagnosis can be made by 2-dimensional echocardiography with Doppler and contrast echocardiography, but the specific diagnosis may require cardiac catheterization and angiocardiography. Treatment is usually performed by a catheter approach. Coils or occlusive devices can be placed relatively safely because most fistulas enter the right atrium or ventricle, so that embolization, if it occurs, is to the lungs, and the coil can easily be retrieved. Care needs to be directed toward not occluding normal coronary arteries. Surgery is occasionally necessary when the fistula is extremely large or proximal.
SYSTEMIC ARTERIOVENOUS FISTULA
Placental arteriovenous fistulas may produce a large increase in fetal cardiac output, particularly in descending aortic blood flow. Although the fistula is no longer present after birth, residual signs may remain in the neonate. These include peripheral edema, cardiomegaly, and a dilated descending aorta.
The most common sites for large arteriovenous communications in children are intracranial, hepatic, or in the extremities. They may be seen as part of Rendu-Osler-Weber syndrome (hereditary hemorrhagic. telangiectasia). They can also be traumatic in origin, most commonly between renal vessels after needle biopsy of the kidney and in the femoral triangle after needling of the femoral vessels.
Because these lesions are obligatory left-to-right shunts, the hemodynamic and clinical features depend on the size of the communication and thus its resistance to flow. The majority of systemic arteriovenous fistulas are small and so do not produce major hemodynamic changes. The exceptions to this are hepatic or intracranial arteriovenous fistulas, particularly those that involve the great vein of Galen or its tributaries. Fistulas can be large and single, multiple, or even resemble cavernous hemangiomas.
Certain clinical features are common to all types of arteriovenous fistulas: a systolic or continuous murmur over the site of the fistula, occasionally a pulsatile mass, and distended and sometimes pulsatile veins draining the region of the fistula. Increased limb size and swelling may occur with peripheral arteriovenous fistulas. Hepatic arteriovenous fistulas generally do not involve one feeder vessel but are usually hemangiomatous.
Intracranial arteriovenous fistulas usually produce the most severe hemodynamic changes because they involve vessels of large caliber and the left-to-right shunt is often large. In early infancy, they may produce severe congestive heart failure, and they are among the few cardiovascular lesions that produce hydrops fetalis or severe congestive failure in the first days after birth. Clinically, they have continuous murmurs over either side of the skull and bounding carotid pulses and distended jugular veins. The superior vena cava is generally markedly dilated on chest radiograph, and there is significant right and left ventricular volume overload. Peripheral pulses are bounding and even collapsing, unless heart failure is so marked that all pulses except the carotids are feeble. If the shunt is not large, cardiovascular manifestations may be mild, and neurologic sequelae dominate the clinical picture. Contrast 2-dimensional echocardiography with Doppler is helpful in diagnosis, but the definitive diagnosis of these lesions may require cardiac catheterization and angiocardiography. Currently arterial embolization is the treatment of choice, but significant neurological sequelae often remain.
Hepatic arteriovenous fistulas may present with congestive heart failure, but like most other hemangiomatous lesions tend to involute in the first year. Involution may be facilitated by giving steroids. Conservative or medical management is appropriate for many of them, especially because the lesions may extend diffusely throughout the liver. If, however, they have uncontrollable congestive heart failure, then the surgeon can either ligate the hepatic artery or, if the lesion is localized, perform a lobectomy.
VENTRICULAR SEPTAL DEFECT
Congenital defects of the interventricular septum are the most common of all congenital heart lesions, accounting for approximately 30% to 60% of all full-term patients with congenital heart malformations; this percentage is equivalent to 3 to 6 of every 1000 live births. This excludes the 3% to 5% of neonates with tiny muscular ventricular septal defects that usually close within the first year. A ventricular septal defect usually occurs as an isolated abnormality but may be associated with other congenital cardiac malformations. In view of the pattern of blood flow in the heart and great vessels of a fetus with a ventricular septal defect, with diversion of blood from the aortic isthmus, narrowing of the aortic isthmus or true coarctation should always be considered when an infant with a ventricular septal defect has severe heart failure. Ventricular septal defects are also common in corrected transposition of the great arteries, in which systemic atrioventricular valve regurgitation and complete heart block are also frequent. They are always present in a truncus arteriosus communis and in a double-outlet right ventricle that, in the absence of pulmonic stenosis, has the clinical features of an isolated ventricular septal defect.
An isolated ventricular septal defect may occur anywhere in the interventricular septum. At birth, about 90% of these defects occur in the muscular septum, but because these usually close spontaneously within 6 to 12 months of birth, the membranous septum becomes the most common site after infancy. Defects vary in size from minute openings to almost complete absence of the interventricular septum (a common ventricle). Most muscular (except multiple, “Swiss cheese”) and perimembranous defects have a high chance of spontaneous closure, unlike large inlet subtricuspid defects, subarterial outlet defects (subaortic as in tetralogy of Fallot or large subpulmonic (as in “supracristal” defects), or doubly committed subarterial ventricular septal defects. Spontaneous partial closure of subpulmonic or doubly committed subarterial defects often involves prolapse of the aortic valve cusp into the defect with development of aortic regurgitation; this form of defect occurs in 5% of whites but in about 35% of Japanese and Chinese. Spontaneous closure of perimembranous defects often is associated with ventricular septal pseudoaneurysm formation; early detection of such an aneurysm indicates a high likelihood of closure.
 CLINICAL MANIFESTATIONS
CLINICAL MANIFESTATIONS
The pathophysiology of left-to-right shunting through a ventricular septal defect involves left and right ventricular volume overloads because the extra volume of the left-to-right shunt passes into the right ventricle before passing into the pulmonary artery.
The systolic murmur of a ventricular septal defect is generally harsh and of the regurgitant or plateau type. With a small shunt, the murmur may be heard only in early systole; as the shunt increases, however, the murmur becomes holosystolic and ends at the aortic component of the second sound. The intensity of the murmur is not necessarily related to the size of the defect, and loud murmurs may be heard with hemodynamically insignificant defects (maladie de Roger). Loud murmurs are usually associated with systolic thrills. The murmur is generally heard best at the lower left sternal border, and it radiates throughout the precordium, but maximally toward the subxiphoid area. However, with a high subpulmonic ventricular septal defect, the maximal intensity may be at the middle to upper left sternal border, with radiation to the right of the sternum. Occasionally the murmur of a very small defect has a crescendo-decrescendo high-pitched almost whistling quality and must be distinguished from an innocent murmur. When the left-to-right shunt is large enough to produce a ratio of pulmonary flow to systemic flow higher than 2:1, a mid-diastolic rumbling murmur may be audible at and inside the apex, and a third sound may appear. As the shunt increases, so does precordial activity. The peripheral arterial pulses give important information about shunt size. If there is a very large shunt that is well above the cardiac output, the left ventricle has to contract forcefully and rapidly to eject the increased stroke volume during a normal systolic interval. This is perceived in the radial and brachial arteries. Unlike in the patent ductus arteriosus, the volume of blood entering the aorta at each stroke is normal, and in diastole there is no rapid runoff through the ductus and less peripheral vasodilatation because baroreceptors are not stimulated. Thus, the rapid fall to a low diastolic pressure is not seen with a ventricular septal defect, but the rapid rise of the pulse gives the impression of a collapsing pulse.
If the defect is small or medium in size, there is no pulmonary hypertension, and the pulmonic component of the second sound is either of normal or minimally increased intensity. If there is pulmonary hypertension, the pulmonic component of the second sound is accentuated. With a small or moderate-sized shunt, the chest roentgenogram shows no or slight increase in left ventricular and left atrial size and pulmonary vascular markings. As the volume of shunting increases, cardiac enlargement and pulmonary vascularity also increase, and pulmonary edema may be seen. Because the shunt is at the ventricular level, the ascending aorta is not dilated. The electrocardiogram is normal if the defect is small; it shows increasing left ventricular hypertrophy as the left-to-right shunt increases, and when there is much right ventricular hypertension, right ventricular hypertrophy is added. A 2-dimensional echocardiogram can be used to show the size and position of the ventricular septal defect. Doppler with imaging techniques can localize the defect by detecting disturbed flow in the right ventricle, and color Doppler flow mapping can demonstrate single or even multiple defects. The Doppler measurement also allows measurement of the pressure gradient across the defect; the higher the gradient, the smaller the defect. In the most severe form of ventricular septal defect, single ventricle complex, magnetic resonance imaging may help to delineate the anatomy. 
 MANAGEMENT
MANAGEMENT
Isolated ventricular septal defects are the most common types of congenital heart disease, so that all pediatricians need to know how they can be managed. The decision tree is shown in Figure 484-1, and the circled numbers in the table are as follows:
1. About 3% to 5% of all live-born babies have small muscular ventricular septal defects, most of which close spontaneously within the next 6 to 12 months. It is neither practical nor reasonable to obtain echocardio-grams in all of them, provided that there appears to be nothing more than a small ventricular septal defect, the heart is quiet, and there are no symptoms. Note that a neonate with a large ventricular septal defect usually has no murmur in the newborn nursery; a large defect with a small shunt across it because of a high pulmonary vascular resistance produces little turbulence. In fact, a typical ventricular septal defect murmur heard in the newborn nursery is almost certainly caused by a small defect.
2. Because defects in the perimembranous or muscular portions of the septum have a high incidence of spontaneous closure, it is appropriate to treat them medically for up to 1 year in the hope that surgery can be averted. When the defect is getting smaller, the systolic murmur may first increase in intensity, but with progressive decrease in size, the murmur becomes softer, and when the defect is extremely small, the murmur becomes shorter and acquires a crescendo-decrescendo high-pitched whistling quality that often portends complete closure. Spontaneous closure may eventually occur in up to 70% of patients, and many of these closures occur by 3 years of age. Most of the closures occurred with small hemodynamically insignificant defects, but even the largest defects can close spontaneously. In a further 25%, the defect becomes smaller but may not close completely; however, the hemodynamic effects are significantly reduced. Because of these statistics, if the defect seems to be becoming smaller, surgical correction should be delayed in the hope of spontaneous closure.
Figure 484-1 also shows reasons for considering early surgery without waiting for the defect to close spontaneously.
3. Some patients respond well enough to medical treatment to go home, but return a week or so later in more severe congestive heart failure. Treatment regimens and doses are adjusted, they improve, and they go home, but the cycle is repeated. These children should be regarded as treatment failures, and they require closure of the defect. Children with trisomy 21 appear to get early pulmonary vascular disease, so their surgery should not be deferred if the defect remains large.
4. Severe social problems are rare reasons for early surgery. These include inability of the parents to bring the child for frequent medical supervision because of distance from the doctor or negligence. In addition, some of these infants are very difficult to manage. They require 2-hourly feeds and consume so much attention that other children in the family are neglected; marriages may even be threatened.
5. Although all infants with large ventricular septal defects grow poorly, with weights usually below the fifth percentile and heights below the 10th percentile, catch-up growth usually occurs once the defect is closed (spontaneously or after surgery). In most of these infants, the growth of head circumference is normal, but in a few head growth falls off rapidly by 3 or 4 months of age. Head growth will return to normal if the defect is closed at this time but fails to catch up if surgery is delayed more than 1 to 2 years.
6. If the patient does not need early surgery for one of the reasons mentioned above, it is appropriate to wait for about 12 months in the hope that the defect will close or become smaller.
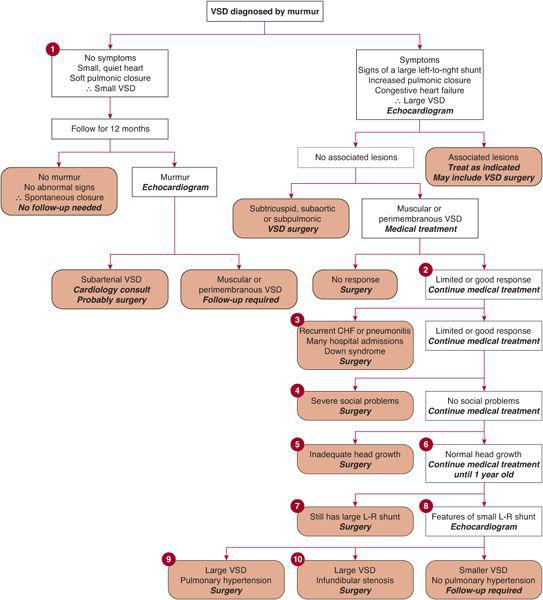
FIGURE 484-1. Decision tree for management of ventricular septal defect (VSD).
7. If the shunt remains large after 1 year of age, there has to be a reason for not closing a large ventricular septal defect because of the increasing risk of irreversible pulmonary vascular disease. By 2 years of age, about 33% of these children have irreversible pulmonary vascular disease.
8. If the left-to-right shunt becomes smaller, there will be clinical improvement, manifested by decreasing cardiac hyperactivity and heart size, diminishing intensity and eventual disappearance of the mid-diastolic murmur, decreasing intensity and changing character of the systolic murmur, lessening and then disappearance of tachypnea, improved appetite and growth, and lessening demand for drug therapy. It is crucial not to be misled into thinking that this improvement necessarily indicates a smaller VSD, because it might also reflect the development of pulmonary vascular disease or, less often, infundibular stenosis. Echocardiography and perhaps cardiac catheterization are mandatory to make decisions about future management at this stage.
9. In most patients with a ventricular septal defect, severe pulmonary vascular disease does not occur until after 1 year of age. However, it can occur earlier, and this will be indicated by a decrease in the left-to-right shunt, a finding that indicates the need for further studies. If obstructive pulmonary vascular disease occurs, there is often little or no left-to-right shunting and no significant right-to-left shunting for several years. (See Chapter 492) However, generally by 5 to 6 years of age, there is increasing cyanosis, particularly during exercise (Eisenmenger syndrome). As severe pulmonary hypertension develops, the main pulmonary artery segment becomes markedly dilated, and the peripheral pulmonary vascular markings on the chest roentgenogram decrease. Obstructive pulmonary vascular disease may progress rapidly in some infants and become irreversible by the age of 12 to 18 months; this should never be allowed to occur. Any doubt as to the cause of any change in clinical status should be investigated by 2-dimensional echocardiography with Doppler or if necessary by cardiac catheterization, and there is good reason to consider routinely recatheterizing children with large ventricular septal defects at 9 to 12 months of age to detect early pulmonary vascular disease that is not clinically apparent. 
10. Infundibular hypertrophy generally develops fairly rapidly, and there may be only a short period in which the left-to-right shunting is present. Soon thereafter, there will be cyanosis, initially on exercise only but then persistently, and the features of the tetralogy of Fallot can develop. In those infants who develop right ventricular outflow obstruction, the incidence of spontaneous closure of a ventricular septal defect is low; a right-to-left shunt can be further complicated by cerebral thrombosis, embolism, or abscess, and the development of infundibular hypertrophy leads to more difficult surgical repair, so that closure of the defect and infundibular resection, if necessary, should be considered early.
Primary surgical closure of the defects can be done with very low mortality. If primary closure is not feasible because of multiple muscular defects or other complicating factors, then banding the pulmonary artery will decrease the left-to-right shunt, reduce pulmonary arterial blood flow and pressure, and relieve congestive heart failure. Banding has its own complications, and removal of the band when the defect is closed later adds to the mortality of the procedure.
Muscular defects, especially if multiple, lead to difficult surgery. From a right ventriculotomy, the masses of hypertrophied trabeculae are daunting and make the defect(s) difficult to find. Although a left ventriculotomy simplifies surgery, a large incision in the systemic ventricle should be avoided. Some surgeons cut away all the right ventricular trabeculae to make closing the defect easier, and others suture all the trabeculae together to close the exit holes. Because of the difficult surgery, closing the muscular defect by catheter introduction of an Amplatzer device is being used more often. Some catheterization procedures are very lengthy, and an alternative is to use a hybrid method in which a surgeon performs a small thoracotomy and the Amplatzer device is inserted more directly through a trocar. Some cardiologists have even used similar devices for nonsurgical closure of perimembranous ventricular septal defects, but this procedure has more risk of producing complete atrioventricular block and of damaging the aortic valve, and it is still considered experimental.
 CONSEQUENCE AND COMPLICATIONS
CONSEQUENCE AND COMPLICATIONS
In several infants with significant reductions in left-to-right shunts caused by spontaneous closing of the ventricular septal defects, mid-to late systolic clicks have become audible. In these children, aneurysmal dilatation of the thin membranous septum or tricuspid valve tissue that has grown to close the defect has occurred, with bulging of pseudoaneurysm into the right ventricle. A small opening often present at the apex of the pseudoaneurysm allows a small left-to-right shunt. Normally, the defect closes, and the pseudoaneurysm slowly shrinks, but rarely it may enlarge progressively. These pseudoaneurysms can be demonstrated by echocardiography.
A number of infants have developed progressive aortic insufficiency associated with ventricular septal defect, particularly if it is subarterial. There is prolapse of an aortic valve leaflet with dilatation of the aortic valve sinus, and rupture of the aortic sinus or cusp may occur. The development of insufficiency has been attributed to stress on the unsupported aortic valve cusp and perhaps suction on it by the jet of the shunt passing through the defect. Even with a small ventricular septal defect, or one showing evidence of closure, aortic insufficiency requires surgical closure of the defect to prevent further prolapse. It may in fact be prudent to close subarterial ventricular septal defects even before evidence of aortic valve cusp involvement is apparent.
Infective endocarditis is an additional problem; rarely, it can occur even after spontaneous closure of the defect. If infective endocarditis involves the tricuspid leaflet sealing the ventricular septal defect, rupture may occur and produce a direct left-ventricular-to-right-atrial communication. Previously, antibiotic prophylaxis for infective endocarditis had been recommended for children with even small defects. Recently, however, the guidelines have been changed and prophylaxis is not recommended except for special circumstances, as discussed in Chapter 490.
L-TRANSPOSITION OF THE GREAT ARTERIES
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


